
NASCarD (Nanopore Adaptive Sampling with Carrier DNA): A Rapid, PCR-Free Method for SARS-CoV-2 Whole-Genome Sequencing in Clinical Samples



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection, RNA Extraction, and cDNA Synthesis
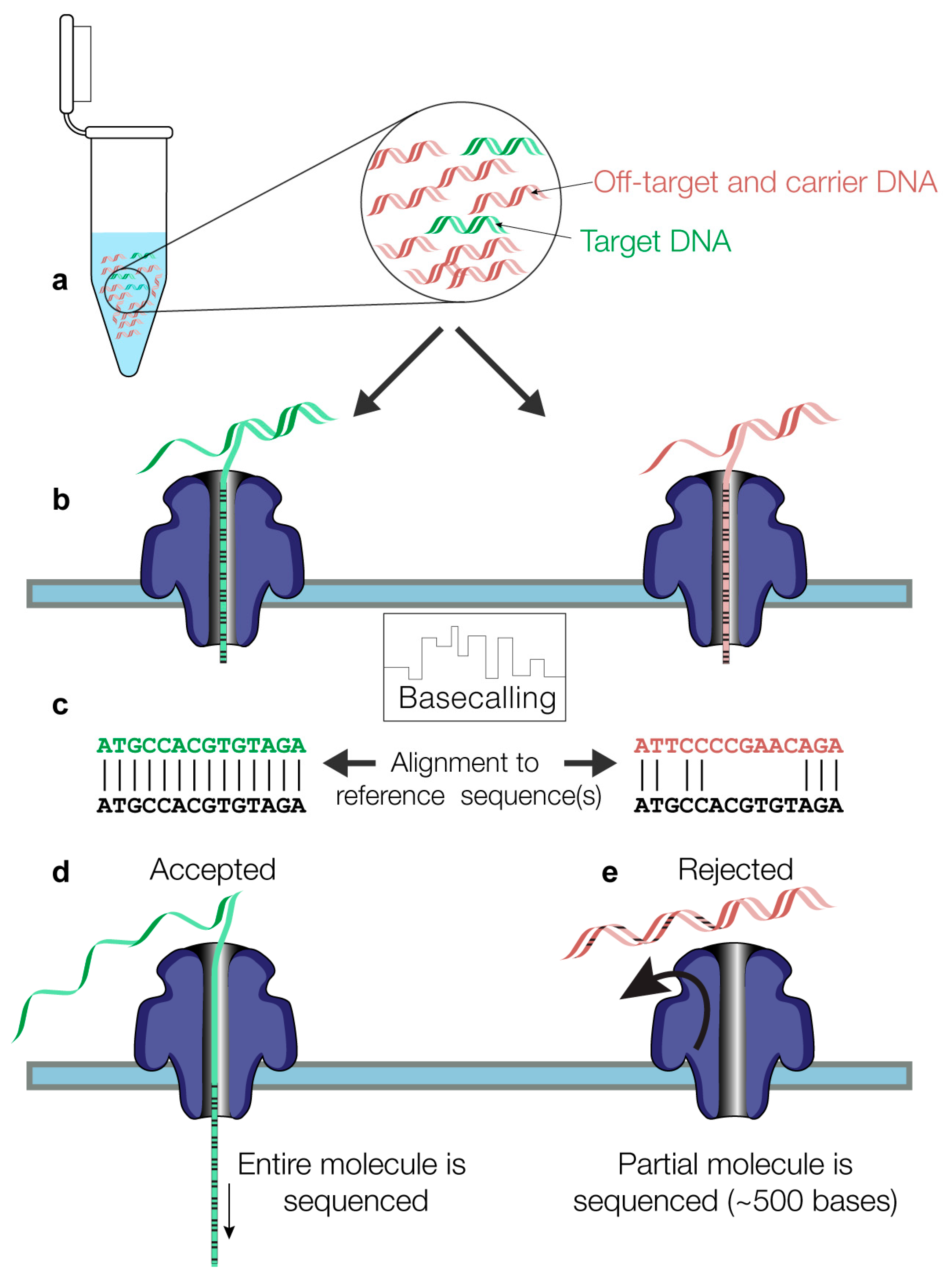
2.2. Nanopore Sequencing
2.3. Bioinformatics Analysis
3. Results
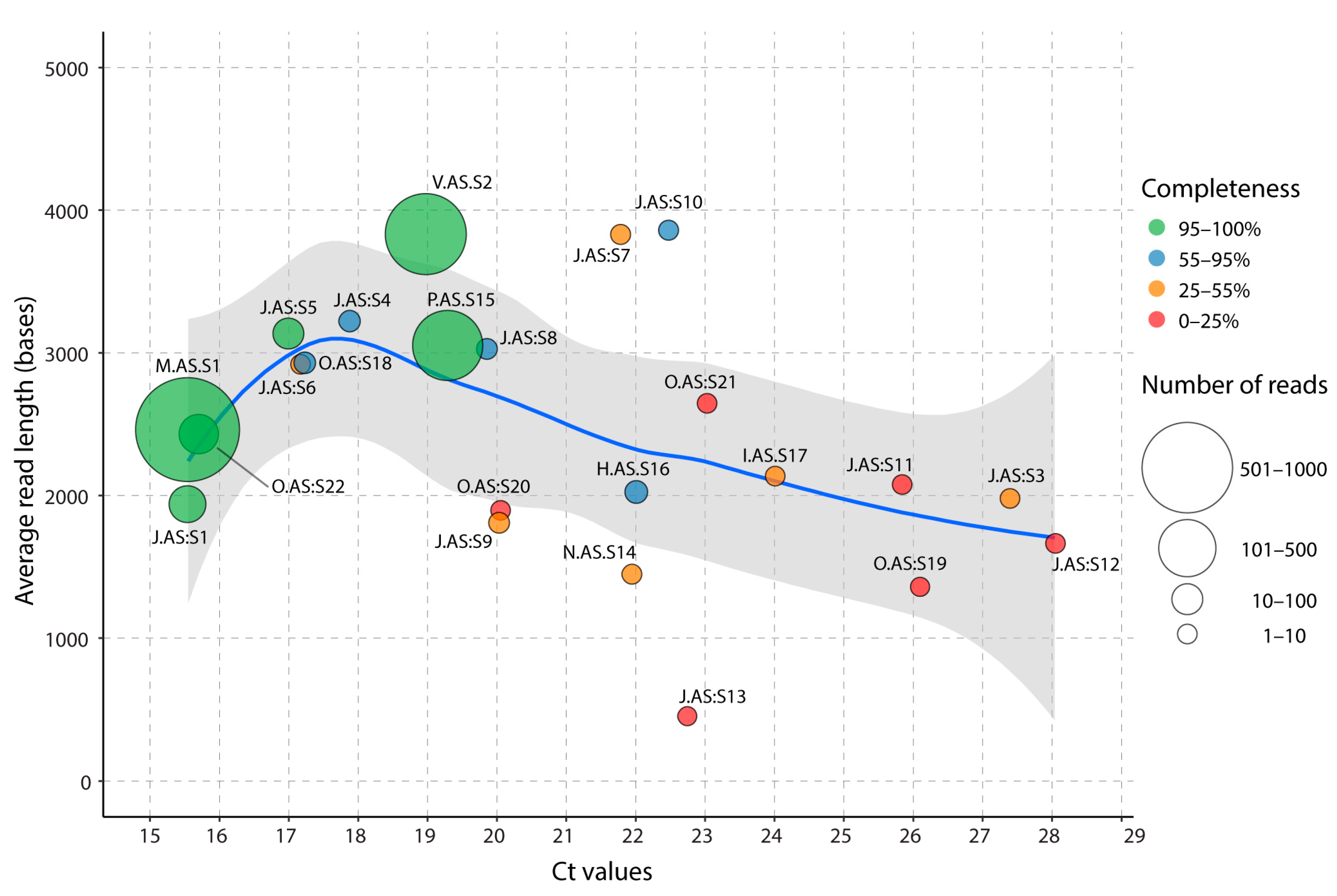
3.1. Increased Yields of SARS-CoV-2 Reads and Bases with NASCarD Lead to Higher Genome Coverage
3.2. Seven Hours Suffice to Obtain High-Quality Consensus Sequences with NASCarD
3.3. GCS Quality Decreases with Lower Viral Loads
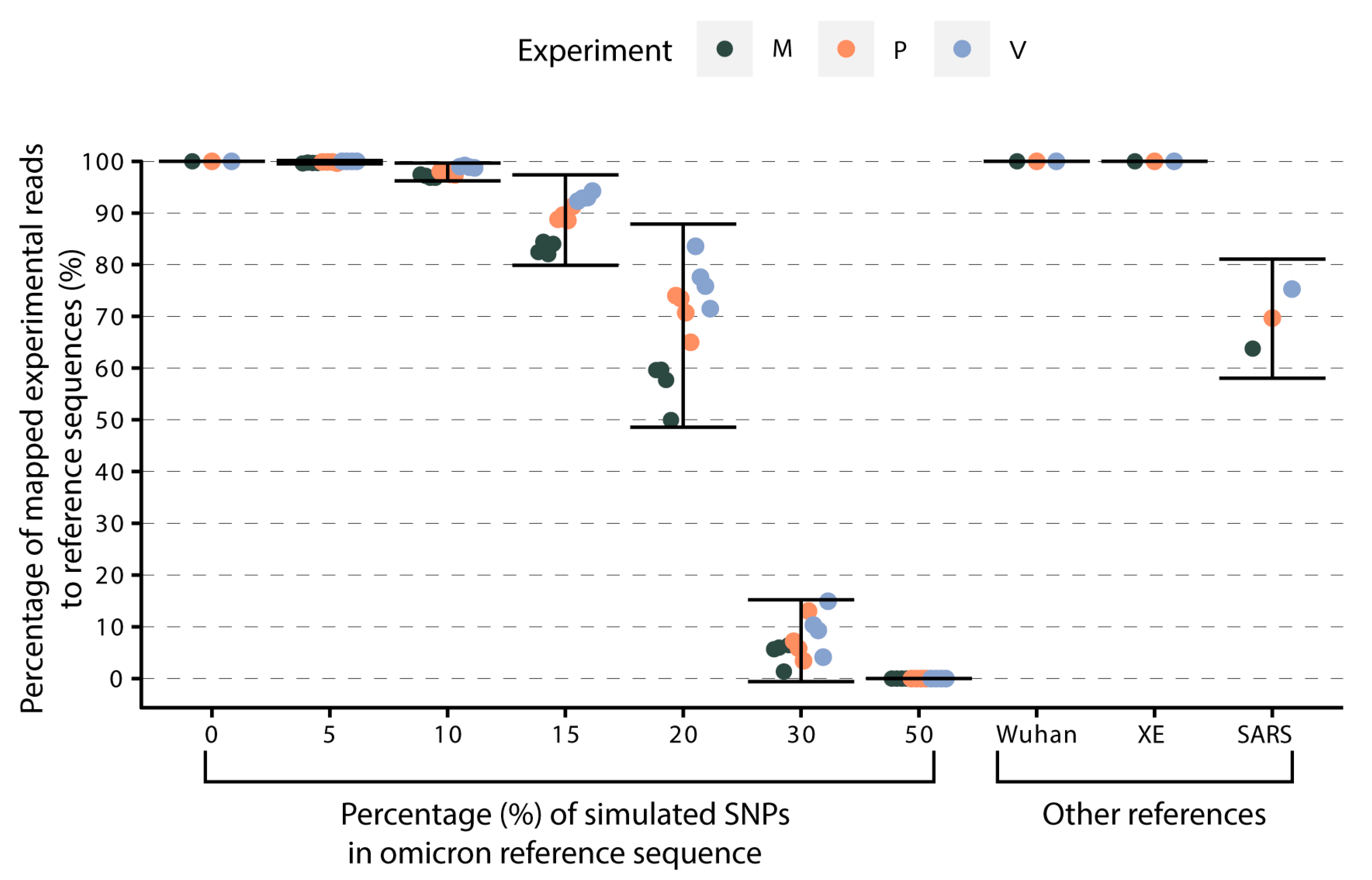
3.4. Effect of the Genetic Divergence of the Reference Sequences on Target Genome Recovery
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Somerville, M.; Curran, J.A.; Dol, J.; Boulos, L.; Saxinger, L.; Doroshenko, A.; Hastings, S.; Reynolds, B.; Gallant, A.J.; Shin, H.D.; et al. Public health implications of SARS-CoV-2 variants of concern: A rapid scoping review. BMJ Open 2021, 11, e055781. [Google Scholar] [CrossRef] [PubMed]
- Vogels, C.B.F.; Breban, M.I.; Ott, I.M.; Alpert, T.; Petrone, M.E.; Watkins, A.E.; Kalinich, C.C.; Earnest, R.; Rothman, J.E.; Goes de Jesus, J.; et al. Multiplex qPCR discriminates variants of concern to enhance global surveillance of SARS-CoV-2. PLoS Biol. 2021, 19, e3001236. [Google Scholar] [CrossRef]
- Bedotto, M.; Fournier, P.E.; Houhamdi, L.; Levasseur, A.; Delerce, J.; Pinault, L.; Padane, A.; Chamieh, A.; Tissot-Dupont, H.; Brouqui, P.; et al. Implementation of an in-house real-time reverse transcription-PCR assay for the rapid detection of the SARS-CoV-2 Marseille-4 variant. J. Clin. Virol. 2021, 139, 104814. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Chen, Z.; Chen, W.; Chen, X.; Hosseini, M.; Yang, Z.; Li, J.; Ho, D.; Turay, D.; Gheorghe, C.P.; et al. A benchmarking study of SARS-CoV-2 whole-genome sequencing protocols using COVID-19 patient samples. iScience 2021, 24, 102892. [Google Scholar] [CrossRef]
- Freed, N.E.; Vlková, M.; Faisal, M.B.; Silander, O.K. Rapid and inexpensive whole-genome sequencing of SARS-CoV-2 using 1200 bp tiled amplicons and Oxford Nanopore Rapid Barcoding. Biol. Methods Protoc. 2020, 5, bpaa014. [Google Scholar] [CrossRef]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef]
- Plitnick, J.; Griesemer, S.; Lasek-Nesselquist, E.; Singh, N.; Lamson, D.M.; St George, K. Whole-Genome Sequencing of SARS-CoV-2: Assessment of the Ion Torrent AmpliSeq Panel and Comparison with the Illumina MiSeq ARTIC Protocol. J. Clin. Microbiol. 2021, 59, e0064921. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Charre, C.; Ginevra, C.; Sabatier, M.; Regue, H.; Destras, G.; Brun, S.; Burfin, G.; Scholtes, C.; Morfin, F.; Valette, M.; et al. Evaluation of NGS-based approaches for SARS-CoV-2 whole genome characterisation. Virus Evol. 2020, 6, veaa075. [Google Scholar] [CrossRef] [PubMed]
- Vrancken, B.; Trovão, N.; Baele, G.; Van Wijngaerden, E.; Vandamme, A.M.; Van Laethem, K.; Lemey, P. Quantifying Next Generation Sequencing Sample Pre-Processing Bias in HIV-1 Complete Genome Sequencing. Viruses 2016, 8, 12. [Google Scholar] [CrossRef]
- Kuchinski, K.S.; Nguyen, J.; Lee, T.D.; Hickman, R.; Jassem, A.N.; Hoang, L.M.N.; Prystajecky, N.A.; Tyson, J.R. Mutations in emerging variant of concern lineages disrupt genomic sequencing of SARS-CoV-2 clinical specimens. Int. J. Infect. Dis. 2022, 114, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Borcard, L.; Gempeler, S.; Terrazos Miani, M.A.; Baumann, C.; Grädel, C.; Dijkman, R.; Suter-Riniker, F.; Leib, S.L.; Bittel, P.; Neuenschwander, S.; et al. Investigating the Extent of Primer Dropout in SARS-CoV-2 Genome Sequences during the Early Circulation of Delta Variants. Front. Virol. 2022, 2, 840952. [Google Scholar] [CrossRef]
- Davis, J.J.; Long, S.W.; Christensen, P.A.; Olsen, R.J.; Olson, R.; Shukla, M.; Subedi, S.; Stevens, R.; Musser, J.M. Analysis of the ARTIC Version 3 and Version 4 SARS-CoV-2 Primers and Their Impact on the Detection of the G142D Amino Acid Substitution in the Spike Protein. Microbiol. Spectr. 2021, 9, e0180321. [Google Scholar] [CrossRef] [PubMed]
- Lambisia, A.W.; Mohammed, K.S.; Makori, T.O.; Ndwiga, L.; Mburu, M.W.; Morobe, J.M.; Moraa, E.O.; Musyoki, J.; Murunga, N.; Mwangi, J.N.; et al. Optimization of the SARS-CoV-2 ARTIC Network V4 Primers and Whole Genome Sequencing Protocol. Front. Med. 2022, 9, 836728. [Google Scholar] [CrossRef] [PubMed]
- Chrzastek, K.; Tennakoon, C.; Bialy, D.; Freimanis, G.; Flannery, J.; Shelton, H. A random priming amplification method for whole genome sequencing of SARS-CoV-2 virus. BMC Genom. 2022, 23, 406. [Google Scholar] [CrossRef]
- Chrzastek, K.; Lee, D.H.; Smith, D.; Sharma, P.; Suarez, D.L.; Pantin-Jackwood, M.; Kapczynski, D.R. Use of Sequence-Independent, Single-Primer-Amplification (SISPA) for rapid detection, identification, and characterization of avian RNA viruses. Virology 2017, 509, 159–166. [Google Scholar] [CrossRef]
- Marotz, C.A.; Sanders, J.G.; Zuniga, C.; Zaramela, L.S.; Knight, R.; Zengler, K. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome 2018, 6, 42. [Google Scholar] [CrossRef]
- Viehweger, A.; Krautwurst, S.; Lamkiewicz, K.; Madhugiri, R.; Ziebuhr, J.; Hölzer, M.; Marz, M. Direct RNA nanopore sequencing of full-length coronavirus genomes provides novel insights into structural variants and enables modification analysis. Genome Res. 2019, 29, 1545–1554. [Google Scholar] [CrossRef]
- Grädel, C.; Terrazos Miani, M.A.; Baumann, C.; Barbani, M.T.; Neuenschwander, S.; Leib, S.L.; Suter-Riniker, F.; Ramette, A. Whole-Genome Sequencing of Human Enteroviruses from Clinical Samples by Nanopore Direct RNA Sequencing. Viruses 2020, 12, 841. [Google Scholar] [CrossRef]
- Vacca, D.; Fiannaca, A.; Tramuto, F.; Cancila, V.; La Paglia, L.; Mazzucco, W.; Gulino, A.; Rosa, M.L.; Maida, C.M.; Morello, G.; et al. Direct RNA Nanopore Sequencing of SARS-CoV-2 Extracted from Critical Material from Swabs. Life 2022, 12, 69. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.M.; Martin, I.W.; Moschetti, W.E.; Kershaw, C.M.; Tsongalis, G.J. Third-Generation Sequencing in the Clinical Laboratory: Exploring the Advantages and Challenges of Nanopore Sequencing. J. Clin. Microbiol. 2019, 58, e01315-19. [Google Scholar] [CrossRef]
- Neuenschwander, S.M.; Terrazos Miani, M.A.; Amlang, H.; Perroulaz, C.; Bittel, P.; Casanova, C.; Droz, S.; Flandrois, J.-P.; Leib, S.L.; Suter-Riniker, F.; et al. A Sample-to-Report Solution for Taxonomic Identification of Cultured Bacteria in the Clinical Setting Based on Nanopore Sequencing. J. Clin. Microbiol. 2020, 58, 1110–1128. [Google Scholar] [CrossRef]
- Loose, M.; Malla, S.; Stout, M. Real-time selective sequencing using nanopore technology. Nat. Methods 2016, 13, 751–754. [Google Scholar] [CrossRef]
- Oxford_Nanopore_Technologies. Read Until-API. 2020. Available online: https://github.com/nanoporetech/read_until_api (accessed on 3 March 2023).
- Kovaka, S.; Fan, Y.; Ni, B.; Timp, W.; Schatz, M.C. Targeted nanopore sequencing by real-time mapping of raw electrical signal with UNCALLED. Nat. Biotechnol. 2020, 39, 431–441. [Google Scholar] [CrossRef]
- Payne, A.; Holmes, N.; Clarke, T.; Munro, R.; Debebe, B.J.; Loose, M. Readfish enables targeted nanopore sequencing of gigabase-sized genomes. Nat. Biotechnol. 2021, 39, 442–450. [Google Scholar] [CrossRef]
- Kipp, E.J.; Lindsey, L.L.; Khoo, B.S.; Faulk, C.D.; Oliver, J.D.; Larsen, P.A. Enabling metagenomic surveillance for bacterial tick-borne pathogens using nanopore sequencing with adaptive sampling. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lin, Y.; Dai, Y.; Liu, Y.; Ren, Z.; Guo, H.; Li, Z.; Li, J.; Wang, K.; Yang, L.; Zhang, S.; et al. Rapid PCR-Based Nanopore Adaptive Sequencing Improves Sensitivity and Timeliness of Viral Clinical Detection and Genome Surveillance. Front. Microbiol. 2022, 13, 929241. [Google Scholar] [CrossRef]
- Martin, S.; Heavens, D.; Lan, Y.; Horsfield, S.; Clark, M.D.; Leggett, R.M. Nanopore adaptive sampling: A tool for enrichment of low abundance species in metagenomic samples. Genome Biol. 2022, 23, 11. [Google Scholar] [CrossRef] [PubMed]
- Heavens, D.; Chooneea, D.; Giolai, M.; Cuber, P.; Aanstad, P.; Martin, S.; Alston, M.; Misra, R.; Clark, M.D.; Leggett, R.M. How Low Can You Go? Driving down the DNA Input Requirements for Nanopore Sequencing. bioRxiv. 2021. bioRxiv:2021.10.15.464554. Available online: https://www.biorxiv.org/content/10.1101/2021.10.15.464554v1 (accessed on 18 February 2023).
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.; Rowe, W.; Rambaut, A. nCoV-2019 Novel Coronavirus Bioinformatics Protocol. 2020. Available online: https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html (accessed on 3 March 2023).
- Kühl, M.A.; Stich, B.; Ries, D.C. Mutation-Simulator: Fine-grained simulation of random mutations in any genome. Bioinformatics 2021, 37, 568–569. [Google Scholar] [CrossRef] [PubMed]
- Quick, J. nCoV-2019 Sequencing Protocol v3 (LoCost). 25 August 2020. Available online: https://protocols.io/view/ncov-2019-sequencing-protocol-v3-locost-bh42j8ye (accessed on 3 March 2023).
- Artic-Network/Fieldbioinformatics. Available online: https://github.com/artic-network/fieldbioinformatics/tree/master/artic (accessed on 3 March 2023).
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Mojarro, A.; Hachey, J.; Ruvkun, G.; Zuber, M.T.; Carr, C.E. CarrierSeq: A sequence analysis workflow for low-input nanopore sequencing. BMC Bioinform. 2018, 19, 108. [Google Scholar] [CrossRef]
- Mokili, J.L.; Rohwer, F.; Dutilh, B.E. Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2012, 2, 63–77. [Google Scholar] [CrossRef]
- Quéromès, G.; Destras, G.; Bal, A.; Regue, H.; Burfin, G.; Brun, S.; Fanget, R.; Morfin, F.; Valette, M.; Trouillet-Assant, S.; et al. Characterization of SARS-CoV-2 ORF6 deletion variants detected in a nosocomial cluster during routine genomic surveillance, Lyon, France. Emerg. Microbes Infect. 2021, 10, 167–177. [Google Scholar] [CrossRef]
- Bal, A.; Destras, G.; Gaymard, A.; Bouscambert-Duchamp, M.; Valette, M.; Escuret, V.; Frobert, E.; Billaud, G.; Trouillet-Assant, S.; Cheynet, V.; et al. Molecular characterization of SARS-CoV-2 in the first COVID-19 cluster in France reveals an amino acid deletion in nsp2 (Asp268del). Clin. Microbiol. Infect. 2020, 26, 960–962. [Google Scholar] [CrossRef]
- Wang, M.; Fu, A.; Hu, B.; Tong, Y.; Liu, R.; Liu, Z.; Gu, J.; Xiang, B.; Liu, J.; Jiang, W.; et al. Nanopore Targeted Sequencing for the Accurate and Comprehensive Detection of SARS-CoV-2 and Other Respiratory Viruses. Small 2020, 16, 2002169. [Google Scholar] [CrossRef] [PubMed]
- Marquet, M.; Zöllkau, J.; Pastuschek, J.; Viehweger, A.; Schleußner, E.; Makarewicz, O.; Pletz, M.W.; Ehricht, R.; Brandt, C. Evaluation of microbiome enrichment and host DNA depletion in human vaginal samples using Oxford Nanopore’s adaptive sequencing. Sci. Rep. 2022, 12, 4000. [Google Scholar] [CrossRef] [PubMed]
- Gan, M.; Wu, B.; Yan, G.; Li, G.; Sun, L.; Lu, G.; Zhou, W. Combined nanopore adaptive sequencing and enzyme-based host depletion efficiently enriched microbial sequences and identified missing respiratory pathogens. BMC Genom. 2021, 22, 732. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, S.J.; Zomer, A.L.; O’Grady, J.; Kay, G.L.; Wain, J.; Janecko, N.; Palau, R.; Mather, A.E. Determination and quantification of microbial communities and antimicrobial resistance on food through host DNA-depleted metagenomics. Food Microbiol. 2023, 110, 104162. [Google Scholar] [CrossRef]
- La Scola, B.; Le Bideau, M.; Andreani, J.; Hoang, V.T.; Grimaldier, C.; Colson, P.; Gautret, P.; Raoult, D. Viral RNA load as determined by cell culture as a management tool for discharge of SARS-CoV-2 patients from infectious disease wards. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1059–1061. [Google Scholar] [CrossRef] [PubMed]
- Wölfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Müller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Fauver, J.R.; Petrone, M.E.; Hodcroft, E.B.; Shioda, K.; Ehrlich, H.Y.; Watts, A.G.; Vogels, C.B.; Brito, A.F.; Alpert, T.; Muyombwe, A.; et al. Coast-to-Coast Spread of SARS-CoV-2 during the Early Epidemic in the United States. Cell 2020, 181, 990–996.e5. [Google Scholar] [CrossRef] [PubMed]
- Sahajpal, N.S.; Mondal, A.K.; Njau, A.; Petty, Z.; Chen, J.; Ananth, S.; Ahluwalia, P.; Williams, C.; Ross, T.M.; Chaubey, A.; et al. High-Throughput Next-Generation Sequencing Respiratory Viral Panel: A Diagnostic and Epidemiologic Tool for SARS-CoV-2 and Other Viruses. Viruses 2021, 13, 2063. [Google Scholar] [CrossRef]
- Ulrich, J.U.; Lutfi, A.; Rutzen, K.; Renard, B.Y. ReadBouncer: Precise and scalable adaptive sampling for nanopore sequencing. Bioinformatics 2022, 38 (Suppl. S1), i153–i160. [Google Scholar] [CrossRef]
- Nagy-Szakal, D.; Couto-Rodriguez, M.; Wells, H.L.; Barrows, J.E.; Debieu, M.; Butcher, K.; Chen, S.; Berki, A.; Hager, C.; Boorstein, R.J.; et al. Targeted Hybridization Capture of SARS-CoV-2 and Metagenomics Enables Genetic Variant Discovery and Nasal Microbiome Insights. Microbiol. Spectr. 2021, 9, e00197-21. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terrazos Miani, M.A.; Borcard, L.; Gempeler, S.; Baumann, C.; Bittel, P.; Leib, S.L.; Neuenschwander, S.; Ramette, A. NASCarD (Nanopore Adaptive Sampling with Carrier DNA): A Rapid, PCR-Free Method for SARS-CoV-2 Whole-Genome Sequencing in Clinical Samples. Pathogens 2024, 13, 61. https://doi.org/10.3390/pathogens13010061
Terrazos Miani MA, Borcard L, Gempeler S, Baumann C, Bittel P, Leib SL, Neuenschwander S, Ramette A. NASCarD (Nanopore Adaptive Sampling with Carrier DNA): A Rapid, PCR-Free Method for SARS-CoV-2 Whole-Genome Sequencing in Clinical Samples. Pathogens. 2024; 13(1):61. https://doi.org/10.3390/pathogens13010061
Chicago/Turabian StyleTerrazos Miani, Miguel A., Loïc Borcard, Sonja Gempeler, Christian Baumann, Pascal Bittel, Stephen L. Leib, Stefan Neuenschwander, and Alban Ramette. 2024. "NASCarD (Nanopore Adaptive Sampling with Carrier DNA): A Rapid, PCR-Free Method for SARS-CoV-2 Whole-Genome Sequencing in Clinical Samples" Pathogens 13, no. 1: 61. https://doi.org/10.3390/pathogens13010061
APA StyleTerrazos Miani, M. A., Borcard, L., Gempeler, S., Baumann, C., Bittel, P., Leib, S. L., Neuenschwander, S., & Ramette, A. (2024). NASCarD (Nanopore Adaptive Sampling with Carrier DNA): A Rapid, PCR-Free Method for SARS-CoV-2 Whole-Genome Sequencing in Clinical Samples. Pathogens, 13(1), 61. https://doi.org/10.3390/pathogens13010061