Abstract
Many types of RNA viruses, including the hepatitis C virus (HCV), activate autophagy in infected cells to promote viral growth and counteract the host defense response. Autophagy acts as a catabolic pathway in which unnecessary materials are removed via the lysosome, thus maintaining cellular homeostasis. The HCV non-structural 5A (NS5A) protein is a phosphoprotein required for viral RNA replication, virion assembly, and the determination of interferon (IFN) sensitivity. Recently, increasing evidence has shown that HCV NS5A can induce autophagy to promote mitochondrial turnover and the degradation of hepatocyte nuclear factor 1 alpha (HNF-1α) and diacylglycerol acyltransferase 1 (DGAT1). In this review, we summarize recent progress in understanding the detailed mechanism by which HCV NS5A triggers autophagy, and outline the physiological significance of the balance between host–virus interactions.
1. Introduction
Infection with RNA viruses, such as HCV, activates host cellular autophagy to promote viral growth and facilitate escape from antiviral defenses [1,2,3,4,5,6,7]. However, how HCV infections regulate autophagy remains controversial. Several HCV viral proteins, such as nonstructural (NS) 5A, may induce autophagy [8,9,10], but their functional roles in regulating autophagy are still unclear. Autophagy is an evolutionarily degradative process that eliminates unnecessary cytosolic materials via lysosomal acidic hydrolases [11,12,13,14,15,16,17]. The precise and proper control of autophagy is critical to maintain cellular homeostasis and ensure the recycling of nutrients [11,12,13,17,18,19,20,21,22]. Chronic HCV infection is the leading cause of late-stage liver disease [19,23,24,25,26,27]. Although current direct-acting antiviral drugs (DAAs) used for treating HCV infection are curative in more than 90% of patients [19,28,29,30,31], the detailed molecular mechanism by which HCV infection induces the development of chronic liver diseases remains unclear. Autophagy is categorized into three types: macroautophagy [11,12,17,32], chaperone-mediated autophagy (CMA) [33,34,35], and microautophagy [36,37,38,39,40]. All of these types of autophagy are critical for regulating the turnover of intracellular components, and their deregulation may develop human diseases [16,20,32]. Interestingly, HCV NS5A protein was recently shown to induce host mitophagy, CMA, and microautophagy, thus eliminating mitochondria, HNF-1α, and DGAT1 [8,9,10], implying that HCV NS5A could be a key player in the regulation of mitochondrial physiology and cellular metabolism in infected cells. In this review, we outline the current knowledge of how HCV NS5A regulates autophagy to promote mitochondrial turnover and protein degradation. Additionally, the regulatory mechanisms involved in these processes are summarized. Finally, the physiological significance and future directions of HCV NS5A-induced autophagy are discussed.
2. HCV
2.1. HCV Epidemiology
Chronic HCV infection in patients is a risk factor for late-stage liver diseases, which ultimately develop into hepatocellular carcinoma (HCC) [19,23,24,25,26,27]. Over 3% of the human population has been infected by HCV, and most of infected individuals progress to chronic liver diseases, generating a global public health burden [19,23,24,25,26,27]. A standard treatment regimen consisting of pegylated interferon-α and ribavirin has been used to control HCV infection for more than twenty years; however, the side effects and low efficacy of this regimen lead to treatment failure in infected individuals [41,42,43]. In the early 2010s, DAAs against HCV genome-encoded protease, phosphoprotein, and RNA-dependent RNA polymerase were developed and approved for the clinical treatment of HCV; these DAAs are curative in approximately 90% of patients [19,28,29,30,31,43]. However, the successful eradication of HCV via treatment with DAAs in infected individuals is hampered by high costs and the emergence of drug-resistant variants after treatment [14,21,28,30,44,45,46].
2.2. Organization of the HCV Genome
HCV is an RNA virus containing an envelope and belongs to the Hepacivirus genus of Flaviviridae family [19,23,25]. The HCV genome comprises a single-stranded, positive-sense RNA with a length of approximately 9.6 kilobases (Kb) [25,47,48]. The RNA genome of HCV is composed of a single open reading frame (ORF) that is flanked by 5′ and 3′ untranslated regions (UTRs) [47,48] (Figure 1). The HCV 5′-UTR contains an internal ribosome entry site (IRES) and a microRNA 122 (miR122)-binding site for activating translation and genome replication [25,47,48,49]. The 3′-UTR of HCV RNA increases IRES-mediated translation and harbors pathogen-associated molecular patterns (PAMPs) that trigger an innate antiviral response [50,51]. The HCV ORF encodes a single polyprotein of approximately 3000 amino acids (a.a.) [47,48] (Figure 1). The translated polypeptide is cleaved by cellular and viral proteases, generating three structural proteins (core, envelope glycoprotein 1 [E1], E2, and P7 proteins) and seven NS proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B proteins) [47] (Figure 1). The structural proteins are responsible for the assembly of infectious HCV particles, whereas the NS proteins participate in HCV replication and assembly of the HCV virion [47] (Figure 1).
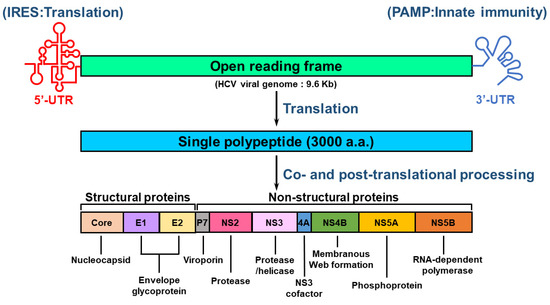
Figure 1.
Genome organization of HCV. The viral genome of HCV consists of a positive-sense, single-stranded RNA with a length of approximately 9.6 kb. The viral genome of HCV includes an open reading frame (ORF) that is flanked by 5′- and 3′-untranslated regions (UTRs). The 5′-UTR contains an internal ribosome entry site (IRES) for stimulating translation, whereas the 3′-UTR harbors a pathogen-associated molecular pattern (PAMP) for inducing innate antiviral immunity. The ORF encodes a single polypeptide that is processed by proteases encoded by host cellular RNA and viral RNA to form structural proteins (core, E1, and E2) and nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B). The core, E1, and E2 proteins are the major constituents of the HCV virion, and other viral proteins differentially regulate the replication of viral RNA and the assembly of infectious particles. The enzymatic activities of these viral proteins are indicated.
2.3. Life Cycle of HCV
HCV primarily infects human hepatocytes and relies on several entry cofactors on the plasma membrane [52,53,54,55,56,57,58,59]. Circulating HCV in the bloodstream associates with lipoproteins, forming lipo-viral particles (LVPs) [60], which initially bind attachment factors on the cell surface, including low-density lipoprotein receptors (LDLRs) [56], scavenger receptor class B member I (SR-BI) [61], and heparan sulfate proteoglycans (HSPGs) [62]. Later, the HCV E2 protein directly interacts with SR-BI and the tetraspanin CD81 [61,63], leading to epidermal growth factor receptor (EGFR)-mediated activation of HRas [57,64]. HRas-induced actin rearrangement then promotes the interaction of CD81 with claudin 1 (CLDN1) [53,65], facilitating HCV internalization via clathrin-mediated endocytosis [66,67]. In addition, Occludin (OCLN), transferrin receptor 1 (TfR1), and the cholesterol receptor Niemann—Pick C1-like L1 (NPC1L1) function in the uptake and entry of HCV [54,58,59]. HCV internalization within acidic endocytotic vesicles drives low pH-dependent fusion of the HCV envelope with the endosome membrane, allowing the viral RNA genome to be released into the cytoplasm [68,69]. This viral RNA can be used as a template for the translation of viral proteins and replication of viral RNA. Finally, the newly synthesized viral genome and structural proteins assemble into infectious HCV virions, which then egress from cells [68,69,70].
3. HCV NS5A
3.1. The Domain Structure of NS5A
The HCV NS5A protein comprises an amphipathic α-helix (AH) at its N-terminus that associates with intracellular membranes, such as the endoplasmic reticulum, and three domains (domains I, II, and III) that are separated by two low-complexity sequences (LCSs, LCSI and LCSII) [71] (Figure 2). HCV NS5A domain I contains four conserved cysteine (Cys) residues capable of binding a single zinc atom, suggesting that HCV NS5A is a metalloprotein that coordinates zinc, which may regulate HCV RNA replication [72,73] (Figure 2). Unlike NS5A domain I, which is highly structured, domains II and III of HCV NS5A are not intrinsically ordered (Figure 2). NS5A domain II is required to replicate the HCV genome [74,75], whereas NS5A domain III participates in the assembly of the HCV virion, rather than in viral RNA replication [76,77] (Figure 2). The interferon sensitivity-determining region (ISDR) and protein kinase R-binding domain (PKRBD), which are located within the C-terminus of LCSI and the N-terminal region of domain II of NS5A, can regulate the sensitivity of the antiviral IFN response [78,79,80,81] (Figure 2). Putative serine (Ser) residues within LCSI may regulate HCV RNA replication [82,83,84]. The polyproline motifs within LCSII can interact with cellular SH3 domains and regulate viral RNA replication and HCV virion assembly [85].
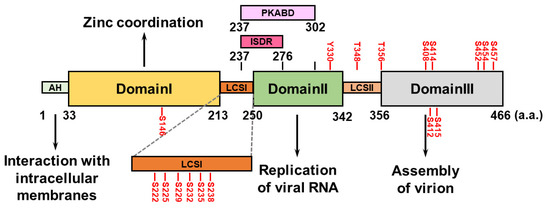
Figure 2.
Domain structure of HCV NS5A. HCV NS5A contains an amphipathic helix (AH) at its N-terminal region and three specific domains: I, II, and III. They are flanked by two low-complexity sequences (LCSs), LCSI and LCSII. The functional role(s) of each domain in the HCV life cycle are specifically indicated. The interferon sensitivity-determining region (ISDR; a.a. 237~276) and the protein kinase R-binding domain (PKRBD; a.a. 237~302) are shown. The serine (S), threonine (T), and tyrosine (Y) residues within HCV NS5A that can be phosphorylated are indicated.
3.2. The Hyperphosphorylation of NS5A
HCV NS5A is a hyperphosphorylated protein containing several Ser and threonine (Thr) residues that can be phosphorylated by intracellular kinases (Figure 2). Phosphorylation of Ser146 in the NS5A domain I negatively regulates the hyperphosphorylated status (p58) of NS5A by reducing the phosphorylation of Ser222 within the LCSI [86]. Phosphorylation at Ser229, Ser232, Ser235, and Ser238 in LCSI is necessary to replicate the HCV genome [82,83,84,87,88]. The phosphorylation of HCV NS5A at Ser225 may facilitate the interaction of NS5A with nucleosome assembly protein 1-like protein 1 (NAP1L1), bridging integrator 1 (Bin1), and vesicle-associated membrane protein-associated protein A (VAPA), thus regulating HCV NS5A localization and viral genome replication [89]. Notably, a recent report revealed that through its ATP-binding domain, HCV NS3A drives the sequential phosphorylation of HCV NS5A at Ser225, Ser232, and Ser235 [90]. The phosphorylation of Thr356, but not Thr348, within LCSII plays a role in HCV RNA replication [86,91]. In addition, ablation of phosphorylation at Ser408, Ser412, Ser414, Ser415, Ser452, Ser454, and 457 within NS5A domain III abolishes infectious particle production [92,93], suggesting that in addition to HCV RNA replication, NS5A phosphorylation regulates HCV assembly. In addition to the Ser and Thr residues, a recent study revealed that tyrosine (Tyr) residue 330 can be phosphorylated, which regulates HCV virion assembly [94]. Several kinases, including cAMP-dependent protein kinase A (PKA), casein kinases I and II (CK-I and CK-II), polo-like kinase (PLK), protein tyrosine kinase, and Abelson murine leukemia viral oncogene homolog 1 (Abl), are responsible for HCV NS5A phosphorylation [84,90,91,94,95,96].
3.3. Functional Role of NS5A in the HCV Life Cycle
Although no specific enzymatic activity of NS5A has been identified, NS5A is considered a multifunctional protein that may regulate HCV RNA replication by binding viral RNA, NS4B, and NS5B [97,98,99,100,101]. Additionally, HCV NS5A can interact with several host cellular factors, including prolyl-peptidyl isomerase, cyclophilin A (CypA) [102,103], VAPA [104,105], phosphatidylinositol-4-kinase IIIα (PI4KIIIα) [106,107], and rab GTP-binding protein 18 (Rab18) [108], to regulate the replication of viral RNA by increasing the binding affinity of NS5A for viral RNA and promoting the formation of a replication compartment, i.e., the “membranous web”. In addition to regulating replication, HCV NS5A may interact with the core to regulate virion production [92]. Moreover, the binding of HCV NS5A to DGAT1 and cortactin facilitates the interaction between HCV NS5A and the core of lipid droplets (LDs) to promote the assembly of infectious particles [109,110]. Since the essential role of NS5A lies in viral RNA replication and assembly of the HCV virion, the suppression of NS5A represents a target for developing DAAs for treating HCV [111,112,113]. Several DAAs against HCV NS5A, such as daclatasvir and ledipasvir, have been used in the clinic to cure HCV [111,112,113]. The persistence of several NS5A resistance-associated substitutions (RASs) in patients after DAA treatment failure significantly reduces the efficacy of DAA treatment [42,46].
4. Autophagy
4.1. Classification of Autophagy
Three forms of autophagy are classified as follows: macroautophagy, microautophagy, and CMA (Figure 3). Macroautophagy (also generally referred to as “autophagy”) involves stepwise vacuole biogenesis to promote the sequestration of intracellular materials within autophagosomes and the delivery of these materials to lysosomes for degradation [11,12,17,32] (Figure 3). Microautophagy involves the invagination and protrusion of the endosome and lysosome membrane to sequester a portion of the cytosol; this process produces intraluminal vesicles in which engulfed materials are degraded [36,37,38,39,40] (Figure 3). Microautophagy can be classified into two forms: fission-type microautophagy, which requires endosomal sorting complexes required for transport (ESCRT)-mediated membrane scission, and fusion-type microautophagy, which involves invagination and extension of the membrane (Figure 3). CMA is a selective process that consists of the recognition of cytosolic cargoes harboring the “Lys-Phe-Glu-Arg-Gln” (KFERQ) pentapeptide motif by the heat shock protein 70 kDa (HSC70, a cytosolic chaperone protein) and the delivery of these cargoes to lysosomes by a lysosomal membrane protein 2A (LAMP2A)-mediated docking process, ultimately leading to the elimination of engulfed cargoes [33,34,35] (Figure 3). Precise control of these three types of autophagy is essential for maintaining cell homeostasis, and their dysregulation may contribute to the development of human diseases [16,17,20,32].
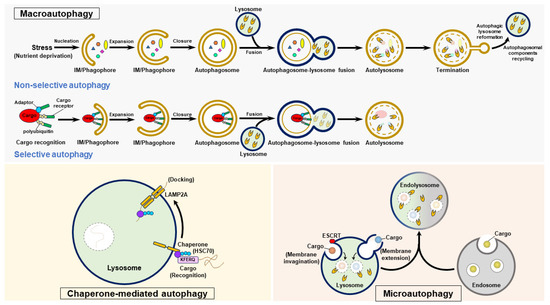
Figure 3.
The autophagy process. Autophagy is categorized into three forms: macroautophagy, chaperone-mediated autophagy (CMA), and microautophagy. To initiate macroautophagy, various stressors, such as nutrient deprivation, induce nucleation of the isolation membrane (IM)/phagophore. The IM/phagophore expands and encloses cytosolic components to form mature autophagosomes surrounded by double membranes. The fusion of autophagosomes with lysosomes generates autolysosomes, and lysosomal hydrolases degrade the elements in the autolysosome interior. The termination of autophagy is coupled with the reformation of tubular lysosome (ALR) and autophagosomal component recycling (ACR), thus regenerating lysosomes and autophagic molecules. Selective autophagy is a specific form of macroautophagy that requires cargo receptors to recognize degradative cargoes via the ubiquitination of cargoes and other adaptor proteins, ultimately delivering these targeted proteins for autophagic degradation. CMA selectively targets degradative substrates harboring the pentapeptide “KFERQ”, which is recognized by HSC70. Through its binding to LAMP2 on the lysosomal membrane, the substrate-HSC70 complex is delivered into lysosomal lumen for degradation. Microautophagy occurs in endosomes and lysosomes, and involves the invagination and protrusion of membrane to sequester cargoes within intraluminal vesicles; these cargoes are then degraded by lysosomal hydrolases.
4.2. Induction of Autophagy
The successful completion of autophagy requires the precise regulation of vacuole biogenesis, and action requires the regular functions of autophagy-related genes (ATGs) and the arrangement of intracellular membranes. The entire process of autophagy includes (1) the generation of a “cup-shaped″ isolation membrane (IM)/phagophore, (2) closure of the IM/phagophores to generate double-membrane autophagosomes, (3) fusion of autophagosomes with lysosomes to generate mature autolysosomes, and (4) lysosome reformation and recycling of autophagosomal components at termination [11,12,13,17,32,114,115,116,117] (Figure 3). Under stress, such as nutrient deficits in mammalian cells, repressed mammalian target of rapamycin complex 1 (mTORC1) activates the unc-51 like-kinase (ULK) complex (ULK1/2, FIP200/RB1CC1, ATG101, and ATG13), inducing the translocation of the ULK complex from the cytosol to the ER-associated membrane [118,119,120,121]. The resident ULK complex at the ER subdomain triggers the activation and recruitment of class III phosphatidylinositol-3-OH kinase (PI3KC3) complex I (PI3KC3/Vps34, PI3KR4/Vps15, ATG14, and Beclin 1), thereby promoting the production of phosphatidylinositol-3-phosphate (PtdIns(3)P) [115,122,123,124]. Newly generated PtdIns(3)P induces the recruitment of double-FYVE-containing protein 1 (DFCP1) and WD-repeat domain PtdIns(3)P-interacting (WIPI) family proteins, promoting the emergence of IM/phagophores (known as cradle-like structures) from the ER via lipid transfer and membrane tethering [115,122,123,125,126]. Other effectors involved in lipid transfer, membrane reconstitution, and IM/phagophore dissociation from the ER also play roles in IM/phagophore formation [127,128,129,130,131,132,133,134]. In addition to the ER, membrane resources from other organelles, including mitochondria, endosomes, the Golgi apparatus, the plasma membrane, and the mitochondria-associated ER membrane (MAM), may also support IM/phagophore formation [135,136,137,138,139,140,141,142].
4.3. Maturation of Autophagosomes
Elongation of the IM/phagophores and their closure to form mature autophagosomes require ubiquitin-like (UBL) conjugation events, including ATG12-ATG5-ATG16 complex conjugation and the attachment of phosphatidylethanolamine (PE) to the ATG8/microtubule-associated protein light chain 3 (LC3) family of proteins covalently (referred to as “ATG8/LC3-PE conjugation” and “ATG8/LC3 lipidation”) [121,143,144,145,146,147]. The enzyme activity of the ubiquitin-like activating enzyme 1 (E1) protein, ATG7 and the ubiquitin-like conjugation enzyme 2 (E2) protein, ATG10 promotes the covalent binding of ATG12 to ATG5, forming an ATG12-ATG5 conjugate. Then, ATG16 binds to ATG12-ATG5, ultimately generating the ATG12-ATG5-ATG16 complex [143,144,148,149,150]. ATG8/LC3 family proteins are posttranslationally processed by ATG4 family protein-mediated cleavage of their C-termini, producing the ATG8/LC3-I form. The conjugation of PE to ATG8/LC3-I subsequently generates ATG8/LC3-II (also known as ATG8/LC3-PE) through the ubiquitin ligase (E3) activity of the ATG12-ATG5-ATG16 complex [151]. Finally, ATG8/LC3-II facilitates membrane fusion to drive IM/phagophore expansion and, ultimately, its enclosure to form double-membrane autophagosomes [146,152,153], in which unwanted intracellular components are sequestered.
4.4. Fusion of Autophagosomes with Lysosomes
Autophagosome—lysosome fusion promotes the formation of autolysosomes, which enable lysosomal acidic hydrolases to degrade engulfed materials, thus promoting the recycling of nutrients [147,154,155,156,157,158]. Several functional molecules involved in vesicle trafficking, membrane tethering, and membrane fusion participate in the autophagosome—lysosome fusion [147,154,155,156,157,158]. Two soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complexes, the syntaxin 17 (STX17)-synaptosome-associated protein 29 (SNAP29)-vesicle-associated membrane protein 7 (VAMP7)/VAMP8 complex [159,160] and the YKT6-SNAP29-STX7 complex [161,162], facilitate membrane fusion. Tethering factors, including the homotypic fusion and protein sorting (HOPS) complex [15], ATG14 [18], and tectonin β-propeller repeat containing 1 (TECPR1) [163] promote the assembly of SNARE complexes during autophagosome—lysosome fusion. Other tethers that similarly function in the fusion of autophagosomes with lysosomes, such as pleckstrin homology domain-containing family M member 1 (PLEKHM1) were also reported recently [15,22,164,165,166,167]. The Rab family of small GTPases [168,169,170,171,172], cytoskeletal motor proteins [171,172,173,174,175,176,177], and cytoskeleton polymerization–mediated minus end-directed and retrograde movement on microtubules [171,178] enable the perinuclear transport of autophagosomes for their fusion with lysosomes.
4.5. Termination of Autophagy
After the process of autophagy is complete, nutrient restoration reactivates mTOR, leading to autophagy termination and autophagic lysosome reformation (ALR) [179,180]. The clathrin-mediated endocytosis regulator (clathrin and adaptor protein 2 [AP2]), the lysosomal efflux permease, spinster, the ubiquitin E3 ligase Cullin 3-Kelch-like protein 20 (KLHL20), and kinesin heavy chain family protein 5B (KIF5B) function in the regulation of ALR. In addition to ALR, autophagosomal component recycling (ACR) was recently shown to function in the termination of autophagy [181,182]. The recycler complex, which is composed of sorting nexins (SNXs, including SNX4, SNX5, and SNX17), promotes the recycling of STX17 and ATG9 via ACR [181,182].
5. Selective Autophagy
5.1. The Process of Selective Autophagy
Autophagy not only serves as a bulk, nonselective degradation process; increasing studies indicate that autophagy may specifically target cargoes, including intracellular organelles and proteins, for degradation, so-called “selective autophagy” [183,184,185,186,187]. The sequestration of degradative cargoes for selective autophagy relies on the recognition of polyubiquitinated proteins by cargo receptors and interactions between cargoes and adaptor proteins [186,187,188,189,190,191,192,193,194] (Figure 3). Many cargo receptors that play a role in selective autophagy, such as p62/sequestosome 1 (p62/SQSTM1), calcium-binding and coiled-coil domain-containing protein 2 (Calcoco2/NDP52), and optineurin (OPTN) have been identified [186,187,190,191,193,194]. The ubiquitin-associated (UBA) domain and LC3-interacting regions (LIRs) within most of these cargo receptors bridge their interaction with ubiquitinated cargoes and binding to ATG8/LC3 family proteins, respectively [186,187,190,191,193,194]. Moreover, two additional GABARAP-interacting motifs and ATG8-interacting motifs within cargo receptors also facilitate the recognition of degradative cargoes in selective autophagy [195,196,197,198].
5.2. Organellophagy
Selective autophagy can eliminate intracellular organelles; this process, referred to as “organellophagy”, involves the removal of damaged organelles and thus facilitates organelle regeneration. Organellophagy can specifically target mitochondria, ER, peroxisomes, lysosomes, ribosomes, nuclei, and LDs for degradation, thereby maintaining organelle integrity [183,185,199]. The regular control of organellophagy promotes organelle regeneration and maintains intracellular metabolic balance.
The removal of deformed mitochondria by organellophagy, called “mitophagy”, represents quality control to regulate mitochondrial integrity [200,201,202]. Mitochondrial depolarization and fission inhibit presenilin-associated rhomboid-like protein (PARL)-mediated cleavage of PTEN-induced putative kinase 1 (PINK1), stabilizing PINK1 on the mitochondrial outer membrane (MOM) [203,204]. The accumulation of PINK1 on the MOM, in turn, induces the recruitment of the ubiquitin E3 ligase Parkin and the phosphorylation of ubiquitin and Parkin at Ser65 [205,206,207,208,209,210]. The translocation of Parkin to mitochondria subsequently triggers the polyubiquitination of MOM proteins [205,206,207,208,211]; this process thus recruits mitophagy receptors, including OPTN and NDP52/Calcoco2, for PINK1/Parkin-dependent mitochondrial turnover [201,212]. In addition, other cargo receptors that analogously drive mitophagy have also been identified recently [213,214,215,216,217,218,219,220,221,222,223,224,225,226]. Moreover, phosphorylation of mitophagy receptors by tank-binding kinase 1 (TBK1) may promote PINK1/Parkin-dependent mitophagy [227,228,229]. Additionally, the recruitment of DFCP1 and WIPI family proteins, the generation of PtdIns(4,5)P2, and F-actin polymerization at the region proximal to degradative mitochondria facilitate IM/phagophore formation for efficient mitochondrial removal via mitophagy [212,230].
6. HCV and Autophagy
6.1. Induction of Autophagy by HCV
HCV was first found to activate autophagy in immortalized HHs (IHHs) harboring the HCV H77 (belonging to genotype 1a) genome [231] (Table 1). Later, the transfection of HCV JFH1 belonging to genotype 2a) RNA in Huh7 human hepatoma cells was shown to induce incomplete autophagy through the unfolded protein response (UPR), and UPR-induced autophagy is required for the replication of HCV [232] (Table 1). The infection of Huh7.5.1 cells with HCV JFH1, a Huh7-derived clone lacking a competent retinoic acid-inducible gene-I (RIG-I) antiviral response, triggers autophagy, which regulates the initial translation of HCV RNA [233,234,235,236] (Table 1). Moreover, the infection of Huh7 cells with HCV JFH1 was demonstrated to activate complete autophagy to suppress HCV PAMP-induced innate antiviral immunity, thus promoting HCV replication [236,237] (Table 1). Similarly, infection of IHHs with HCV H77 also induces autophagy to repress the IFN antiviral response [236,237,238] (Table 1). The inhibition of antiviral immunity by HCV-induced autophagy was further supported by the finding that tumor necrosis factor receptor (TNFR)-associated factor 6 (TRAF6) degradation is induced through the autophagy receptor p62/SQSTM1 in infected cells [239] (Table 1). In addition, HCV-induced autophagy reportedly impairs IFN-α-triggered innate immune signaling by downregulating the level of IFN-α receptor 1(IFNAR1) [240] (Table 1). These findings are also supported by Mori et al.’s study, which revealed that the replication of HCV JFH1 induces selective autophagy in Huh7.5.1 cells [241] (Table 1). In addition, HCV JFH1 infection activates autophagy to promote the turnover of mitochondria, LDs, and viral proteins [9,242,243,244] (Table 1). Mitochondrial degradation by Parkin-dependent mitophagy in HCV-infected cells prevents cell death and promotes persistent virus infection [243,245] (Table 1). Given that the quality control of mitochondria by mitophagy plays a crucial role in controlling innate immunity [246,247,248,249,250], it remains unclear whether HCV-induced mitophagy regulates HCV PAMP-triggered innate antiviral response. Additionally, HCV-induced autophagy could participate in the catabolism of LDs through an unresolved mechanism [242] (Table 1). In addition to innate antiviral immunity, HCV infection has also been shown to activate the nucleotide-binding domain, leucine-rich-containing family, pyrin domain–containing-3 (NLRP3) inflammasome, which leads to the release of interleukin 1 beta (IL-1β) [251,252,253]. Although autophagy and mitophagy have been shown to regulate the activation of the NLRP3 inflammasome [254,255], whether HCV-induced autophagy and mitophagy modulate the NLRP3 inflammasome response remains uncertain. These studies imply that HCV-activated autophagy and mitophagy may interfere with innate antiviral immunity, alter the homeostasis of lipid metabolism, and regulate the inflammatory response.
Several studies have shown that HCV triggers the biogenesis of autophagosomes [256,257,258,259,260]. The induction of autophagosomes by HCV induces the regeneration of membranous compartments to replicate HCV viral RNA [259] (Table 1), mainly through the interaction of ATG5 with NS4B and NS5B transiently [260] (Table 1). Molecules that function in the initial stage of autophagy, including PI3KC3/Vps34 and DFCP1, reportedly positively regulate the replication of viral RNA via the formation of a membranous structure [256] (Table 1). At the initial infection stage, HCV can induce the expression of the run domain Beclin-1-interacting and cysteine-rich domain-containing protein (Rubicon) and repress UV irradiation resistance-associated gene protein (UVRAG) expression to delay the maturation of autophagosomes, which support HCV viral RNA replication [258] (Table 1). In addition, HCV infection induces the emergence of phagophores from the ER and triggers STX7-mediated homotypic fusion for the biogenesis of autophagosomes, thus supporting the replication of HCV viral RNA [257] (Table 1). On the other hand, autophagy regulates the HCV virion assembly in infected cells [261] (Table 1). HCV-induced autophagy may promote the egress of infectious virions through apolipoprotein E (ApoE) transport and the CD63-associated exosome pathway [262,263] (Table 1). These studies indicate that virus-induced autophagy plays a proviral role in the HCV life cycle.

Table 1.
Regulation of autophagy by HCV.
Table 1.
Regulation of autophagy by HCV.
| Approach | Phenotype | Functional Role | Reference(s) |
|---|---|---|---|
| Infection of IHHs with HCV H77 (1a) |
| Promote viral RNA replication | [231] |
| Transfection of HCV JFH1 (2a) RNA in Huh7.5 cells |
| Promote viral RNA replication | [232] |
| Infection of Huh7 cells with HCV JFH1 |
| Promote the translation of incoming viral RNA | [236] |
| Infection of Huh7 cells with HCV-JFH1 |
| Promote viral RNA replication/repress innate antiviral immunity | [237] |
| Infection of IHHs with HCV-H77 and HCV-JFH1 |
| Promote viral RNA replication/repress innate antiviral immunity | [238] |
| Infection of Huh7 cells with HCV-JFH1 |
| Promote viral RNA replication/repress innate antiviral immunity | [239] |
| Infection of Huh7 and Huh7.5 cells with HCV-JFH1 |
| Suppress IFN-α-triggered innate antiviral signaling | [240] |
| HCV-JFH1 infection and HCV JFH1 subgenomic RNA transfection in Huh7 and Huh7.5.1 cells |
| Unclear | [241] |
| Transfection of HCV Con1 (1b) and HCV JFH1 subgenomic RNA in Huh7 and Huh7.5-1 cells |
| Promote LD catabolism | [242] |
| HCV-JFH1 infection and transfection of Con1 subgenomic RNA in Huh7.5.1 cells |
| Protect infected cells from apoptosis/establish viral persistence | [243,245] |
| Transfection of HCV JFH1 RNA in Huh7.5 cells |
| Promote viral RNA replication | [259] |
| Infection of Huh7 cells with HCV-JFH1 |
| Promote viral RNA replication | [260] |
| Transfection of HCV Con1 (1b) and HCV JFH1 subgenomic RNA in Huh7/Infection of Huh7 cells with HCV JC1 (2a) |
| Promote viral RNA replication | [256,259] |
| Infection of Huh7 and Huh7.5 cells with HCV-JFH1 |
| Promote viral RNA replication | [258] |
| Infection of Huh7 and Huh7.5 cells with HCV-JFH1 |
| Promote viral RNA replication | [257] |
| Infection of Huh7.5-1 cells with HCV-JFH1 |
| Enhance the assembly of viral particles | [261] |
| Infection of IHHs and Huh7.5 cells with HCV-H77 and HCV-JFH1 |
| Enhance the assembly of viral particles | [262] |
| Infection of Huh7 and Huh7.5-1 cells with HCV-JFH1 |
| Enhance the assembly of viral particles | [263] |
6.2. Activation of Autophagy by HCV Viral Proteins
Among the HCV genome-encoded proteins, NS4B was the first viral protein shown to induce incomplete autophagy in Huh7 cells through Rab5 and PI3KC3, which may promote HCV replication [264] (Table 2). HCV NS3 can trigger autophagy via an immunity-associated GTPase family M (IRGM)-dependent pathway to increase viral infectivity [265] (Table 2). In addition, the HCV core protein also induces autophagy via the UPR and eukaryotic translation initiation factor 2 alpha kinase 3 (EIF2AK3)-activating transcription factor 6 (ATF6) axis-mediated upregulation of ATG12 and LC3B [266] (Table 2). In contrast, the HCV core protein may repress mitophagy by interfering with Parkin translocation to mitochondria and later mitochondrial ubiquitination [267] (Table 2). Moreover, HCV NS5A also induces autophagy to promote mitochondrial turnover, HNF-1α degradation (Table 2), and DGAT1 degradation [9,10,268] (Table 2). These studies indicate that HCV proteins play specific roles in regulating autophagy.
Table 2.
Induction of autophagy by the HCV core, NS3, and NS4B proteins.
7. Regulation of Autophagy by HCV NS5A
7.1. Induction of Mitophagy by HCV NS5A Eliminates Mitochondria
Siu and colleagues first reported that the transfection of HCV JFH1 RNA in Huh7 cells induces ER–mitochondrion tethering, which is characterized by the formation of a MAM that wraps around the mitochondrion [269] (Table 3). Additionally, the authors showed that ectopic expression of HCV NS5A induces the formation of ER-tethered mitochondria, to which HCV NS5A is mainly localized, in human embryonic kidney 293 (HEK293) cells [269] (Table 3). In addition, the overexpression of HCV NS5A induces mitochondrial fragmentation, regardless of the activity of dynamin-related protein 1 (Drp1) [269] (Table 3). Moreover, PI4KIIIα inhibitor treatment and overexpression of a PI4KIIIα kinase-dead (KD) mutant suppressed mitochondrial fragmentation in HCV NS5A-overexpressing cells [269] (Table 3), suggesting that PI4KIIIα kinase activity is required for HCV NS5A-induced mitochondrial fragmentation. Similarly, interference with the interaction between HCV NS5A and PI4KIIIα by ectopic expression of a PI4KIIIα mutant lacking an NS5A-interacting motif and HCV NS5A lacking the PI4KIIIα-binding domain repressed mitochondrial fragmentation in cells [269] (Table 3), suggesting that the interaction between HCV NS5A and PI4KIIIα is critical for the induction of mitochondrial fragmentation. The overexpression of PI4KIIIα KD and a dominant-negative mutant of Drp1 (K38D) inhibited mitochondrial fragmentation in Huh7.5.1 cells infected with HCV JC1 (belonging to genotype 2a) [269] (Table 3), indicating that both PI4KIIIα and Drp1 are necessary for HCV-induced mitochondrial fragmentation. Furthermore, the overexpression of HCV NS5A in cells alleviated hydrogen peroxide-induced cell apoptosis [269] (Table 3). Together, these studies suggest that HCV NS5A may induce mitochondrial fragmentation through its interaction with PI4KIIIα and that this process could prevent cells from undergoing apoptosis.
Table 3.
Activation of autophagy by HCV NS5A.
Soon afterward, Jassey et al. reported that ectopic expression of HCV NS5A in Huh-7.5 cells induces the formation of green fluorescence protein (GFP)-tagged LC3 puncta and increases the expression of PE-conjugated LC3, resembling the effects of rapamycin and carbonyl cyanide m-chlorophenyl hydrazine (CCCP) [8] (Table 3), suggesting that HCV NS5A expression alone is sufficient to activate autophagy. Staining with the dye JC-1, which acts as an indicator of the mitochondrial membrane potential, coupled with flow cytometry and immunofluorescence analysis revealed that the overexpression of HCV NS5A in Huh-7.5 cells induces depolarization and mitochondrial fragmentation [8] (Table 3), indicating that HCV NS5A expression is sufficient to drive mitochondrial deformation. In addition, biochemical and immunofluorescence analyses revealed that HCV NS5A overexpression triggers the recruitment of Parkin to mitochondria, which is similar to the effects of CCCP treatment [8] (Table 3), suggesting that HCV NS5A promotes mitochondrial translocation. The induced production of mCherry+/EGFP+ autophagosomes and mCherry+/EGFP− autolysosomes containing the mCherry-EGFP-LC3 reporter and the increased accumulation of LC3-II induced by bafilomycin A1 in HCV NS5A-overexpressing cells suggest that HCV NS5A activates complete autophagy [8] (Table 3). Moreover, treatment with the antioxidant N-acetyl cysteine (NAC) attenuated the expression of total LC3-II and mitochondrial LC3-II in Huh-7.5 cells harboring HCV NS5A [8] (Table 3), indicating that suppression of reactive oxygen species (ROS) inhibits HCV NS5A-induced autophagy and mitophagy. Furthermore, overexpression of the HCV core protein in HCV NS5A-overexpressing Huh-7.5 cells diminishes the mitochondrial translocation of Parkin [8], suggesting that the HCV core protein interferes with HCV NS5A-induced mitophagy. These studies imply that HCV NS5A may drive mitophagy and that the HCV core protein may suppress this process. The physiological significance of HCV NS5A-activated mitophagy in the pathogenesis of HCV-related liver diseases remains largely unclear. Mitophagy has been implicated in the development of liver diseases, including HCC [270,271,272,273]. In particular, mitophagy activation was recently shown to promote HCC development by inducing the production of ROS, the stemness of cancer stem cells (CSCs), and the reprogramming of energy metabolism [274,275,276]. Therefore, the functional role(s) of HCV NS5A-induced mitophagy in the occurrence of HCV-associated HCC and its clinical relevance should be investigated in the future. Since mitophagy has emerged its role(s) in regulating viral PAMP-induced antiviral signaling and NLRP3 inflammasome activation, whether HCV NS5A activates mitophagy to modulate innate antiviral immunity and the inflammatory response in HCV-infected cells is interesting to study in the future.
7.2. HCV NS5A-Induced CMA Degrades HNF-1α
Matsui and colleagues reported that HNF-1α is a transcription factor that may transactivate the mRNA expression of glucose transporter 2 (GLUT2) during HCV replication [277]. The overexpression of HCV NS5A during HCV J6/JFH1 (belonging to genotype 2a) infection leads to lysosome-dependent HNF-1α degradation [277]. The degradation of HNF-1α within lysosomes induced by HCV may repress GLUT2 mRNA expression, thus interfering with glucose metabolism [277]. Matsui et al. further reported that ectopically expressed HNF-1α coimmunoprecipitated with endogenous and overexpressed HSC70, the chaperone involved in CMA, in Huh-7.5 cells [9] (Table 3). The authors reported that HNF-1α contains a putative pentapeptide (“Gln-Arg-Glu-Val-Val”) that can interact with the CMA chaperone HSC70 and that mutation of this motif diminished the binding of HNF-1α to HSC70 [9] (Table 3), suggesting that HNF-1α is a substrate of CMA-mediated degradation. The overexpression of HCV NS5A promotes the binding of HNF-1α to HSC70, and HCV JFH1 infection also increases the binding of HNF-1α to HSC70 [9] (Table 3). In addition to HNF-1α, HCV NS5A can bind HSC70 through its domain I (a.a. 121~126) [9] (Table 3), implying that HCV NS5A connects the interaction of HNF-1α with HSC70. Moreover, HCV J6/JFH1 infection leads to HNF-1α degradation, which is inhibited by gene knockdown of LAMP2A and HSC70, and by ammonia chloride treatment [9] (Table 3), suggesting that HCV infection may activate CMA to degrade HNF-1α. Immunofluorescence analysis revealed that HCV J6/JFH1 infection promoted the colocalization of HCV NS5A and HNF-1α [9] (Table 3). Additionally, ectopic expression of HCV NS5A leads to the redistribution of HNF-1α to lysosomes containing HCV NS5A [9] (Table 3), suggesting that HCV NS5A acts as a bridge to localize HNF-1α within lysosomes. Treatment with the lysosomal degradation inhibitor pepstatin A suppresses the lysosomal degradation of HNF-1α, and a mutation in the pentapeptide motif of HNF-1α (Gln130Ala) reduces the lysosomal translocation of HNF-1α [9] (Table 3). These studies indicate that HCV NS5A facilitates the recruitment of HNF-1α to HSC70 in lysosomes, thus inducing HNF-1α degradation via CMA. Somatic mutations in HNF-1α have been found in the cancerous tissues of 1~2% of HCC patients [278], and are highly related to cell stemness and liver malignancies [279], indicating that the HCV NS5A-regulated CMA-mediated HNF-1α could participate in the pathogenesis of HCV-associated HCC. However, further studies are needed to explore the clinical relevance of HCV NS5A-activated CMA in the development of HCC.
7.3. The Triggering of Endosomal Microautophagy by HCV NS5A Induces DGAT1 Degradation
DGAT1 is a critical enzyme that catalyzes the final step in triglyceride biosynthesis, which is necessary for the biogenesis of LDs [280]. Additionally, DGAT1 is the host cellular factor required for the efficient assembly of the HCV virion, presumably through its ability to enhance the interaction between HCV NS5A and the core protein on the LD surface [109,281]. Yuliandari and colleagues reported that ectopically expressed HCV NS5A coimmunoprecipitates with DGAT1, which is overexpressed in Huh-7.5 cells [10] (Table 3). Additionally, immunofluorescence analysis of these overexpressed cells showed that HCV NS5A and DGAT1 colocalize [10] (Table 3). The author reported that HCV NS5A interacts with DGAT1 through domain I of HCV NX5A (a.a. 1-213) and that a.a. 157~399 of DGAT1 are critical for the binding of DGAT1 to HCV NS5A [10] (Table 3). In addition, HCV J6/JFH1 infection leads to DGAT1 degradation, which is inhibited by ammonia chloride [10] (Table 3), suggesting that DGAT1 is a substrate of lysosomal degradation. Similarly, HCV NS5A overexpression also induces the degradation of DGAT1 [10] (Table 3). Moreover, DGAT1 contains a pentapeptide, “Gln-Val-Glu-Lys-Arg” (a.a. 149~153), that can interact with HSC70, a chaperone necessary for CMA and endosomal microautophagy (eMI) [10] (Table 3). A mutation within this pentapeptide (Gln149Ala) of DGAT1 interrupts the binding of DGAT1 to HSC70 [10] (Table 3), suggesting that DGAT1 specifically binds HSC70. The authors found, by immunofluorescence analysis, that DGAT1 colocalizes with HCV NS5A on lysosomes and late endosomes. Genetic knockdown of VPS4B, which functions in eMI but not LAMP2A, restores DGAT1 expression in HCV J6/JFH1-infected cells [10] (Table 3). Collectively, these studies revealed that eMI is the main route by which HCV induces the lysosomal degradation of DGAT1. Since DGAT1 has been shown to regulate LD biogenesis and HCV virion assembly [109,280,281], investigating whether HCV NS5A-induced degradation of DGAT1 via eMI may interfere with the biogenesis of LDs from excess free fatty acids (FFAs) and establish persistent infection in infected cells is valuable. Also, the physiological significance and clinical relevance of HCV NS5A-induced eMI in the pathogenesis of HCV-related liver steatosis require further investigation.
8. Conclusions and Perspectives
Several recent studies have suggested that HCV NS5A plays an unexpected role in regulating HCV-induced autophagy [8,9,10]. Notably, HCV NS5A induces autophagy to selectively eliminate mitochondria via mitophagy and target HNF-1α and DGAT1 for CMA- and eMI-mediated degradation, respectively (Figure 4). Importantly, these studies suggest a new paradigm, in which HCV NS5A alone is sufficient to trigger autophagic degradation, which may allow cells to counteract oxidative stress [8], reduce glucose metabolism [9], and alter LD biogenesis [10] (Figure 4). However, the detailed mechanism by which HCV NS5A induces these autophagy-related degradation processes is largely unknown. In the context of mitophagy, how HCV NS5A induces mitochondrial depolarization and promotes the mitochondrial translocation of Parkin is unclear. Since the HCV core protein has been shown to interrupt mitophagy in cells harboring HCV NS5A [8,273] and to affect the LD localization of HCV NS proteins [282], whether HCV NS5A has a similar phenotype in infected cells in which the complete HCV life cycle is carried out is questionable. Additionally, it remains unclear whether HCV NS5A, which is overexpressed, maintains a competent hyperphosphorylation status similar to that of NS5A, which is encoded by the entire HCV genome in infected cells. Moreover, the specific domain of HCV NS5A and whether the hyperphosphorylation of HCV NS5A can alter the ability of NS5A to activate mitophagy are also unknown. Further studies are urgently needed to determine whether and how the HCV NS5A phosphorylation status regulates mitochondrial turnover in HCV-infected cells.
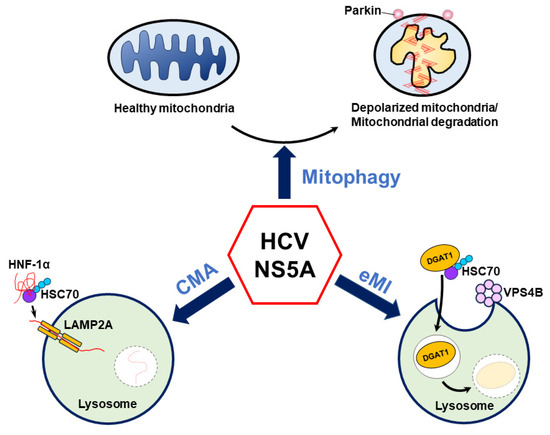
Figure 4.
Current model of HCV NS5A-regulated autophagy. HCV NS5A induces mitochondrial depolarization and fission and triggers mitochondrial translocation to remove deformed mitochondria via mitophagy. Additionally, HCV NS5A acts as a bridge to connect HNF-1α and HSC70 on lysosomes, allowing the degradation of HNF-1α by LAMP2A-mediated CMA. In addition, HCV NS5A promotes the degradation of DGAT1 via endosomal microautophagy (eMI) through the binding of HSC70 and functional VPS4B.
Intriguingly, HCV NS5A is sufficient to activate CMA, which is accompanied mainly by macroautophagy activation in cells. HCV NS5A may direct the interaction of HNF-1α with HSC70 on lysosomes, and this process could differ from the LD-mediated relocalization of HCV NS5A mediated by the capsid protein in HCV-infected cells [282]. However, whether and how HCV NS5A is redistributed into various subcellular compartments to activate mitophagy, CMA, and eMI remain largely unknown. Additionally, little is known about how and why HCV infection simultaneously triggers these diverse autophagy processes through NS5A in infected cells. In this context, further investigations are urgently needed to delineate the underlying mechanism and depict the spatiotemporal control of HCV NS5A-mediated activation of autophagic degradation in cells in which the entire HCV life cycle can be carried out. Domains I and II of HCV NS5A are crucial to the replication of viral RNA [72,73,74,75], and domain III functions in virion assembly [76,77]. Therefore, it is interesting to study whether HCV NS5A may activate autophagy to promote virus replication by reconstituting intracellular membranes in the future. Additionally, whether HCV-induced autophagy regulates the stability and intracellular trafficking of intracellular proteins to enhance the assembly of infectious particles is worth further investigation. Furthermore, additional studies are needed to determine whether HCV NS5A-activated mitophagy, CMA, and eMI regulate HCV growth and participate in the development of HCV-related liver diseases. Also, the physiological significance and clinical relevance of HCV NS5A-induced mitochondrial turnover, CMA-induced degradation of HNF-1α, and eMI-mediated DGAT1 degradation in the pathogenesis of HCV-associated liver diseases, including liver steatosis and HCC, urgently need to be studied. In addition, whether HCV NS5A-induced mitophagy is involved in the regulation of innate antiviral immunity and inflammasome activation, thereby deregulating the immunological response and affecting clinical outcomes in HCV-infected patients, also needs to be understood in the future. On the other hand, it remains uncertain whether treatment with HCV DAAs against NS5A in infected patients suppresses the degradation of mitochondria, HNF-1α, and DGAT1 in HCV-infected hepatocytes. Many abnormal mitochondria and the ER remain in the liver tissues of HCV-infected patients after one year of treatment with DAAs [283], which suggests that pharmacological modulation of mitophagy, CMA, and eMI could be used to improve the clinical consequences of HCV infection after treatment with DAAs.
Author Contributions
Conceptualization: P.-Y.K.; original draft preparation: P.-Y.K.; review: P.-Y.K. and C.-T.Y.; editing: P.-Y.K. and C.-T.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by research grants from the National Science and Technology Council (NSTC 113-2311-B-182-001 and MOST 109-2320-B-182-010-MY3) in Taipei, the National Health Research Institute (NHRI-EX103~106-10322SC) in Miaoli, and Chang Gung Memorial Hospital (CMRPD1P0261, CMRPD1N0431, CMRPD1N0221, and BMRPD01) in Taoyuan, Taiwan.
Acknowledgments
The authors acknowledge the support of research grants from the National Science and Technology Council, National Health Research Institute, and Chang Gung Memorial Hospital. The authors apologize to colleagues whose work could not be cited owing to space-related restraints.
Conflicts of Interest
The authors have no conflicts of interest to declare.
References
- Chan, S.T.; Ou, J.J. Hepatitis C Virus-Induced Autophagy and Host Innate Immune Response. Viruses 2017, 9, 224. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.Y.K.; Ou, J.J. Autophagy in HCV Replication and Protein Trafficking. Int. J. Mol. Sci. 2021, 22, 1089. [Google Scholar] [CrossRef]
- Ke, P.Y. The Multifaceted Roles of Autophagy in Flavivirus-Host Interactions. Int. J. Mol. Sci. 2018, 19, 3940. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.Y.; Chen, S.S. Autophagy in hepatitis C virus-host interactions: Potential roles and therapeutic targets for liver-associated diseases. World J. Gastroenterol. 2014, 20, 5773–5793. [Google Scholar] [CrossRef]
- Ke, P.Y. Autophagy and antiviral defense. IUBMB Life 2022, 74, 317–338. [Google Scholar] [CrossRef]
- Ke, P.Y. Crosstalk between Autophagy and RLR Signaling. Cells 2023, 12, 956. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ou, J.J. HCV-induced autophagy and innate immunity. Front. Immunol. 2024, 15, 1305157. [Google Scholar] [CrossRef]
- Jassey, A.; Liu, C.H.; Changou, C.A.; Richardson, C.D.; Hsu, H.Y.; Lin, L.T. Hepatitis C Virus Non-Structural Protein 5A (NS5A) Disrupts Mitochondrial Dynamics and Induces Mitophagy. Cells 2019, 8, 290. [Google Scholar] [CrossRef]
- Matsui, C.; Deng, L.; Minami, N.; Abe, T.; Koike, K.; Shoji, I. Hepatitis C Virus NS5A Protein Promotes the Lysosomal Degradation of Hepatocyte Nuclear Factor 1alpha via Chaperone-Mediated Autophagy. J. Virol. 2018, 92, e00639-18. [Google Scholar] [CrossRef]
- Yuliandari, P.; Matsui, C.; Deng, L.; Abe, T.; Mori, H.; Taguwa, S.; Ono, C.; Fukuhara, T.; Matsuura, Y.; Shoji, I. Hepatitis C virus NS5A protein promotes the lysosomal degradation of diacylglycerol O-acyltransferase 1 (DGAT1) via endosomal microautophagy. Autophagy Rep. 2022, 1, 264–285. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Morishita, H.; Mizushima, N. Diverse Cellular Roles of Autophagy. Annu. Rev. Cell Dev. Biol. 2019, 35, 453–475. [Google Scholar] [CrossRef]
- Howe, A.Y.M.; Rodrigo, C.; Cunningham, E.B.; Douglas, M.W.; Dietz, J.; Grebely, J.; Popping, S.; Sfalcin, J.A.; Parczewski, M.; Sarrazin, C.; et al. Characteristics of hepatitis C virus resistance in an international cohort after a decade of direct-acting antivirals. JHEP Rep. 2022, 4, 100462. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Nishimura, T.; Sakamaki, Y.; Itakura, E.; Hatta, T.; Natsume, T.; Mizushima, N. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol. Biol. Cell 2014, 25, 1327–1337. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Yamamoto, H.; Zhang, S.; Mizushima, N. Autophagy genes in biology and disease. Nat. Rev. Genet. 2023, 24, 382–400. [Google Scholar] [CrossRef]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef]
- Manns, M.P.; Maasoumy, B. Breakthroughs in hepatitis C research: From discovery to cure. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 533–550. [Google Scholar] [CrossRef]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef]
- Baumert, T.F.; Berg, T.; Lim, J.K.; Nelson, D.R. Status of Direct-Acting Antiviral Therapy for Hepatitis C Virus Infection and Remaining Challenges. Gastroenterology 2019, 156, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Tabata, K.; Matsunaga, K.; Sakane, A.; Sasaki, T.; Noda, T.; Yoshimori, T. Rubicon and PLEKHM1 negatively regulate the endocytic/autophagic pathway via a novel Rab7-binding domain. Mol. Biol. Cell 2010, 21, 4162–4172. [Google Scholar] [CrossRef] [PubMed]
- Chisari, F.V. Unscrambling hepatitis C virus-host interactions. Nature 2005, 436, 930–932. [Google Scholar] [CrossRef]
- Zoulim, F.; Chevallier, M.; Maynard, M.; Trepo, C. Clinical consequences of hepatitis C virus infection. Rev. Med. Virol. 2003, 13, 57–68. [Google Scholar] [CrossRef]
- Manns, M.P.; Buti, M.; Gane, E.; Pawlotsky, J.M.; Razavi, H.; Terrault, N.; Younossi, Z. Hepatitis C virus infection. Nat. Rev. Dis. Primers 2017, 3, 17006. [Google Scholar] [CrossRef]
- Martinello, M.; Solomon, S.S.; Terrault, N.A.; Dore, G.J. Hepatitis C. Lancet 2023, 402, 1085–1096. [Google Scholar] [CrossRef] [PubMed]
- Thrift, A.P.; El-Serag, H.B.; Kanwal, F. Global epidemiology and burden of HCV infection and HCV-related disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 122–132. [Google Scholar] [CrossRef]
- Dore, G.J.; Bajis, S. Hepatitis C virus elimination: Laying the foundation for achieving 2030 targets. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 91–92. [Google Scholar] [CrossRef]
- Gotte, M.; Feld, J.J. Direct-acting antiviral agents for hepatitis C: Structural and mechanistic insights. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 338–351. [Google Scholar] [CrossRef]
- Marshall, A.D.; Willing, A.R.; Kairouz, A.; Cunningham, E.B.; Wheeler, A.; O’Brien, N.; Perera, V.; Ward, J.W.; Hiebert, L.; Degenhardt, L.; et al. Direct-acting antiviral therapies for hepatitis C infection: Global registration, reimbursement, and restrictions. Lancet Gastroenterol. Hepatol. 2024, 9, 366–382. [Google Scholar] [CrossRef]
- Rehermann, B. HCV in 2015: Advances in hepatitis C research and treatment. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, T.; Yoshimori, T. Autophagosome biogenesis and human health. Cell Discov. 2020, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 2011, 7, 673–682. [Google Scholar] [CrossRef]
- Oku, M.; Sakai, Y. Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Machineries. Bioessays 2018, 40, e1800008. [Google Scholar] [CrossRef]
- Schuck, S. Microautophagy—Distinct molecular mechanisms handle cargoes of many sizes. J. Cell Sci. 2020, 133, jcs246322. [Google Scholar] [CrossRef]
- Wang, L.; Klionsky, D.J.; Shen, H.M. The emerging mechanisms and functions of microautophagy. Nat. Rev. Mol. Cell Biol. 2023, 24, 186–203. [Google Scholar] [CrossRef]
- Kuchitsu, Y.; Taguchi, T. Lysosomal microautophagy: An emerging dimension in mammalian autophagy. Trends Cell Biol. 2024, 34, 606–616. [Google Scholar] [CrossRef]
- Berenguer, M. Systematic review of the treatment of established recurrent hepatitis C with pegylated interferon in combination with ribavirin. J. Hepatol. 2008, 49, 274–287. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, M.S.; Cooper, C.; Hunyady, B.; Jia, J.; Ogurtsov, P.; Peck-Radosavljevic, M.; Shiffman, M.L.; Yurdaydin, C.; Dalgard, O. Management of adverse effects of Peg-IFN and ribavirin therapy for hepatitis C. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Hong, F.; Radaeva, S. Host factors and failure of interferon-alpha treatment in hepatitis C virus. Hepatology 2004, 39, 880–890. [Google Scholar] [CrossRef]
- Sarrazin, C. Treatment failure with DAA therapy: Importance of resistance. J. Hepatol. 2021, 74, 1472–1482. [Google Scholar] [CrossRef]
- Li, D.K.; Chung, R.T. Overview of Direct-Acting Antiviral Drugs and Drug Resistance of Hepatitis C Virus. Methods Mol. Biol. 2019, 1911, 3–32. [Google Scholar] [CrossRef]
- Pawlotsky, J.M. Hepatitis C Virus Resistance to Direct-Acting Antiviral Drugs in Interferon-Free Regimens. Gastroenterology 2016, 151, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef]
- Dubuisson, J.; Cosset, F.L. Virology and cell biology of the hepatitis C virus life cycle: An update. J. Hepatol. 2014, 61, S3–S13. [Google Scholar] [CrossRef]
- Panigrahi, M.; Palmer, M.A.; Wilson, J.A. MicroRNA-122 Regulation of HCV Infections: Insights from Studies of miR-122-Independent Replication. Pathogens 2022, 11, 1005. [Google Scholar] [CrossRef]
- Ito, T.; Tahara, S.M.; Lai, M.M. The 3′-untranslated region of hepatitis C virus RNA enhances translation from an internal ribosomal entry site. J. Virol. 1998, 72, 8789–8796. [Google Scholar] [CrossRef]
- Saito, T.; Owen, D.M.; Jiang, F.; Marcotrigiano, J.; Gale, M., Jr. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 2008, 454, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Vitelli, A.; Granier, C.; Goujon, C.; Dubuisson, J.; Pascale, S.; Scarselli, E.; Cortese, R.; Nicosia, A.; Cosset, F.L. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem. 2003, 278, 41624–41630. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wolk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Ploss, A.; Evans, M.J.; Gaysinskaya, V.A.; Panis, M.; You, H.; de Jong, Y.P.; Rice, C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef]
- Zhang, J.; Randall, G.; Higginbottom, A.; Monk, P.; Rice, C.M.; McKeating, J.A. CD81 is required for hepatitis C virus glycoprotein-mediated viral infection. J. Virol. 2004, 78, 1448–1455. [Google Scholar] [CrossRef]
- Agnello, V.; Abel, G.; Elfahal, M.; Knight, G.B.; Zhang, Q.X. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 12766–12771. [Google Scholar] [CrossRef]
- Lupberger, J.; Zeisel, M.B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.J.; Turek, M.; Gorke, S.; et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011, 17, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B., Jr.; Barretto, N.; Martin, D.N.; Hiraga, N.; Imamura, M.; Hussain, S.; Marsh, K.A.; Yu, X.; Chayama, K.; Alrefai, W.A.; et al. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 2012, 18, 281–285. [Google Scholar] [CrossRef]
- Martin, D.N.; Uprichard, S.L. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc. Natl. Acad. Sci. USA 2013, 110, 10777–10782. [Google Scholar] [CrossRef]
- Andre, P.; Komurian-Pradel, F.; Deforges, S.; Perret, M.; Berland, J.L.; Sodoyer, M.; Pol, S.; Brechot, C.; Paranhos-Baccala, G.; Lotteau, V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 2002, 76, 6919–6928. [Google Scholar] [CrossRef]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Schafer, C.; Adah, M.I.; Zhang, F.; Linhardt, R.J.; Toyoda, H.; Kinoshita-Toyoda, A.; Toida, T.; Van Kuppevelt, T.H.; Depla, E.; et al. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 2003, 278, 41003–41012. [Google Scholar] [CrossRef] [PubMed]
- Pileri, P.; Uematsu, Y.; Campagnoli, S.; Galli, G.; Falugi, F.; Petracca, R.; Weiner, A.J.; Houghton, M.; Rosa, D.; Grandi, G.; et al. Binding of hepatitis C virus to CD81. Science 1998, 282, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Zona, L.; Lupberger, J.; Sidahmed-Adrar, N.; Thumann, C.; Harris, H.J.; Barnes, A.; Florentin, J.; Tawar, R.G.; Xiao, F.; Turek, M.; et al. HRas signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell Host Microbe 2013, 13, 302–313. [Google Scholar] [CrossRef]
- Harris, H.J.; Farquhar, M.J.; Mee, C.J.; Davis, C.; Reynolds, G.M.; Jennings, A.; Hu, K.; Yuan, F.; Deng, H.; Hubscher, S.G.; et al. CD81 and claudin 1 coreceptor association: Role in hepatitis C virus entry. J. Virol. 2008, 82, 5007–5020. [Google Scholar] [CrossRef]
- Blanchard, E.; Belouzard, S.; Goueslain, L.; Wakita, T.; Dubuisson, J.; Wychowski, C.; Rouille, Y. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J. Virol. 2006, 80, 6964–6972. [Google Scholar] [CrossRef]
- Farquhar, M.J.; Hu, K.; Harris, H.J.; Davis, C.; Brimacombe, C.L.; Fletcher, S.J.; Baumert, T.F.; Rappoport, J.Z.; Balfe, P.; McKeating, J.A. Hepatitis C virus induces CD81 and claudin-1 endocytosis. J. Virol. 2012, 86, 4305–4316. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Rice, C.M. The ins and outs of hepatitis C virus entry and assembly. Nat. Rev. Microbiol. 2013, 11, 688–700. [Google Scholar] [CrossRef]
- Jones, D.M.; McLauchlan, J. Hepatitis C virus: Assembly and release of virus particles. J. Biol. Chem. 2010, 285, 22733–22739. [Google Scholar] [CrossRef]
- Paul, D.; Madan, V.; Bartenschlager, R. Hepatitis C virus RNA replication and assembly: Living on the fat of the land. Cell Host Microbe 2014, 16, 569–579. [Google Scholar] [CrossRef]
- Ross-Thriepland, D.; Harris, M. Hepatitis C virus NS5A: Enigmatic but still promiscuous 10 years on! J. Gen. Virol. 2015, 96, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Tellinghuisen, T.L.; Marcotrigiano, J.; Gorbalenya, A.E.; Rice, C.M. The NS5A protein of hepatitis C virus is a zinc metalloprotein. J. Biol. Chem. 2004, 279, 48576–48587. [Google Scholar] [CrossRef] [PubMed]
- Tellinghuisen, T.L.; Marcotrigiano, J.; Rice, C.M. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 2005, 435, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Tellinghuisen, T.L.; Foss, K.L.; Treadaway, J.C.; Rice, C.M. Identification of residues required for RNA replication in domains II and III of the hepatitis C virus NS5A protein. J. Virol. 2008, 82, 1073–1083. [Google Scholar] [CrossRef]
- Ross-Thriepland, D.; Amako, Y.; Harris, M. The C terminus of NS5A domain II is a key determinant of hepatitis C virus genome replication, but is not required for virion assembly and release. J. Gen. Virol. 2013, 94, 1009–1018. [Google Scholar] [CrossRef]
- Appel, N.; Zayas, M.; Miller, S.; Krijnse-Locker, J.; Schaller, T.; Friebe, P.; Kallis, S.; Engel, U.; Bartenschlager, R. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 2008, 4, e1000035. [Google Scholar] [CrossRef]
- Hughes, M.; Griffin, S.; Harris, M. Domain III of NS5A contributes to both RNA replication and assembly of hepatitis C virus particles. J. Gen. Virol. 2009, 90, 1329–1334. [Google Scholar] [CrossRef]
- Paterson, M.; Laxton, C.D.; Thomas, H.C.; Ackrill, A.M.; Foster, G.R. Hepatitis C virus NS5A protein inhibits interferon antiviral activity, but the effects do not correlate with clinical response. Gastroenterology 1999, 117, 1187–1197. [Google Scholar] [CrossRef]
- Song, J.; Fujii, M.; Wang, F.; Itoh, M.; Hotta, H. The NS5A protein of hepatitis C virus partially inhibits the antiviral activity of interferon. J. Gen. Virol. 1999, 80 Pt 4, 879–886. [Google Scholar] [CrossRef]
- Gale, M., Jr.; Blakely, C.M.; Kwieciszewski, B.; Tan, S.L.; Dossett, M.; Tang, N.M.; Korth, M.J.; Polyak, S.J.; Gretch, D.R.; Katze, M.G. Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: Molecular mechanisms of kinase regulation. Mol. Cell Biol. 1998, 18, 5208–5218. [Google Scholar] [CrossRef]
- Enomoto, N.; Sakuma, I.; Asahina, Y.; Kurosaki, M.; Murakami, T.; Yamamoto, C.; Izumi, N.; Marumo, F.; Sato, C. Comparison of full-length sequences of interferon-sensitive and resistant hepatitis C virus 1b. Sensitivity to interferon is conferred by amino acid substitutions in the NS5A region. J. Clin. Investig. 1995, 96, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Appel, N.; Pietschmann, T.; Bartenschlager, R. Mutational analysis of hepatitis C virus nonstructural protein 5A: Potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J. Virol. 2005, 79, 3187–3194. [Google Scholar] [CrossRef]
- Lemay, K.L.; Treadaway, J.; Angulo, I.; Tellinghuisen, T.L. A hepatitis C virus NS5A phosphorylation site that regulates RNA replication. J. Virol. 2013, 87, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Matsunaga, S.; Takahashi, H.; Nakashima, K.; Kimura, Y.; Ito, M.; Matsuda, M.; Murayama, A.; Kato, T.; Hirano, H.; et al. Involvement of hepatitis C virus NS5A hyperphosphorylation mediated by casein kinase I-alpha in infectious virus production. J. Virol. 2014, 88, 7541–7555. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.; Gretton, S.; Shelton, H.; Brown, D.D.; McCormick, C.J.; Angus, A.G.; Patel, A.H.; Griffin, S.; Harris, M. A conserved proline between domains II and III of hepatitis C virus NS5A influences both RNA replication and virus assembly. J. Virol. 2009, 83, 10788–10796. [Google Scholar] [CrossRef]
- Ross-Thriepland, D.; Harris, M. Insights into the complexity and functionality of hepatitis C virus NS5A phosphorylation. J. Virol. 2014, 88, 1421–1432. [Google Scholar] [CrossRef]
- Chong, W.M.; Hsu, S.C.; Kao, W.T.; Lo, C.W.; Lee, K.Y.; Shao, J.S.; Chen, Y.H.; Chang, J.; Chen, S.S.; Yu, M.J. Phosphoproteomics Identified an NS5A Phosphorylation Site Involved in Hepatitis C Virus Replication. J. Biol. Chem. 2016, 291, 3918–3931. [Google Scholar] [CrossRef]
- Eyre, N.S.; Hampton-Smith, R.J.; Aloia, A.L.; Eddes, J.S.; Simpson, K.J.; Hoffmann, P.; Beard, M.R. Phosphorylation of NS5A Serine-235 is essential to hepatitis C virus RNA replication and normal replication compartment formation. Virology 2016, 491, 27–44. [Google Scholar] [CrossRef]
- Goonawardane, N.; Gebhardt, A.; Bartlett, C.; Pichlmair, A.; Harris, M. Phosphorylation of Serine 225 in Hepatitis C Virus NS5A Regulates Protein-Protein Interactions. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Yu, C.C.; Lin, P.C.; Chiang, C.H.; Jen, S.T.; Lai, Y.L.; Hsu, S.C.; Lo, L.C.; Lin, J.J.; Chan, N.L.; Yu, M.J. Sequential Phosphorylation of Hepatitis C Virus NS5A Protein Requires the ATP-Binding Domain of NS3 Helicase. J. Virol. 2022, 96, e0010722. [Google Scholar] [CrossRef]
- Cordek, D.G.; Croom-Perez, T.J.; Hwang, J.; Hargittai, M.R.; Subba-Reddy, C.V.; Han, Q.; Lodeiro, M.F.; Ning, G.; McCrory, T.S.; Arnold, J.J.; et al. Expanding the proteome of an RNA virus by phosphorylation of an intrinsically disordered viral protein. J. Biol. Chem. 2014, 289, 24397–24416. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Suzuki, R.; Murakami, K.; Aizaki, H.; Ishii, K.; Murayama, A.; Date, T.; Matsuura, Y.; Miyamura, T.; Wakita, T.; et al. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 2008, 82, 7964–7976. [Google Scholar] [CrossRef] [PubMed]
- Tellinghuisen, T.L.; Foss, K.L.; Treadaway, J. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 2008, 4, e1000032. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, D.; Takeuchi, K.; Chihara, K.; Fujieda, S.; Sada, K. Protein tyrosine kinase Abl promotes hepatitis C virus particle assembly via interaction with viral substrate activator NS5A. J. Biol. Chem. 2022, 298, 101804. [Google Scholar] [CrossRef]
- Chen, Y.C.; Su, W.C.; Huang, J.Y.; Chao, T.C.; Jeng, K.S.; Machida, K.; Lai, M.M. Polo-like kinase 1 is involved in hepatitis C virus replication by hyperphosphorylating NS5A. J. Virol. 2010, 84, 7983–7993. [Google Scholar] [CrossRef]
- Quintavalle, M.; Sambucini, S.; Summa, V.; Orsatti, L.; Talamo, F.; De Francesco, R.; Neddermann, P. Hepatitis C virus NS5A is a direct substrate of casein kinase I-alpha, a cellular kinase identified by inhibitor affinity chromatography using specific NS5A hyperphosphorylation inhibitors. J. Biol. Chem. 2007, 282, 5536–5544. [Google Scholar] [CrossRef]
- Huang, L.; Hwang, J.; Sharma, S.D.; Hargittai, M.R.; Chen, Y.; Arnold, J.J.; Raney, K.D.; Cameron, C.E. Hepatitis C virus nonstructural protein 5A (NS5A) is an RNA-binding protein. J. Biol. Chem. 2005, 280, 36417–36428. [Google Scholar] [CrossRef]
- Foster, T.L.; Belyaeva, T.; Stonehouse, N.J.; Pearson, A.R.; Harris, M. All three domains of the hepatitis C virus nonstructural NS5A protein contribute to RNA binding. J. Virol. 2010, 84, 9267–9277. [Google Scholar] [CrossRef]
- Shirota, Y.; Luo, H.; Qin, W.; Kaneko, S.; Yamashita, T.; Kobayashi, K.; Murakami, S. Hepatitis C virus (HCV) NS5A binds RNA-dependent RNA polymerase (RdRP) NS5B and modulates RNA-dependent RNA polymerase activity. J. Biol. Chem. 2002, 277, 11149–11155. [Google Scholar] [CrossRef]
- Quezada, E.M.; Kane, C.M. The Stimulatory Mechanism of Hepatitis C Virus NS5A Protein on the NS5B Catalyzed Replication Reaction In Vitro. Open Biochem. J. 2013, 7, 11–14. [Google Scholar] [CrossRef]
- David, N.; Yaffe, Y.; Hagoel, L.; Elazar, M.; Glenn, J.S.; Hirschberg, K.; Sklan, E.H. The interaction between the hepatitis C proteins NS4B and NS5A is involved in viral replication. Virology 2015, 475, 139–149. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hanoulle, X.; Badillo, A.; Wieruszeski, J.M.; Verdegem, D.; Landrieu, I.; Bartenschlager, R.; Penin, F.; Lippens, G. Hepatitis C virus NS5A protein is a substrate for the peptidyl-prolyl cis/trans isomerase activity of cyclophilins A and B. J. Biol. Chem. 2009, 284, 13589–13601. [Google Scholar] [CrossRef]
- Chatterji, U.; Lim, P.; Bobardt, M.D.; Wieland, S.; Cordek, D.G.; Vuagniaux, G.; Chisari, F.; Cameron, C.E.; Targett-Adams, P.; Parkinson, T.; et al. HCV resistance to cyclosporin A does not correlate with a resistance of the NS5A-cyclophilin A interaction to cyclophilin inhibitors. J. Hepatol. 2010, 53, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Gao, L.; Shi, S.T.; Taylor, D.R.; Yang, T.; Mircheff, A.K.; Wen, Y.; Gorbalenya, A.E.; Hwang, S.B.; Lai, M.M. Hepatitis C virus RNA polymerase and NS5A complex with a SNARE-like protein. Virology 1999, 263, 30–41. [Google Scholar] [CrossRef]
- Hamamoto, I.; Nishimura, Y.; Okamoto, T.; Aizaki, H.; Liu, M.; Mori, Y.; Abe, T.; Suzuki, T.; Lai, M.M.; Miyamura, T.; et al. Human VAP-B is involved in hepatitis C virus replication through interaction with NS5A and NS5B. J. Virol. 2005, 79, 13473–13482. [Google Scholar] [CrossRef]
- Reiss, S.; Rebhan, I.; Backes, P.; Romero-Brey, I.; Erfle, H.; Matula, P.; Kaderali, L.; Poenisch, M.; Blankenburg, H.; Hiet, M.S.; et al. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 2011, 9, 32–45. [Google Scholar] [CrossRef]
- Reiss, S.; Harak, C.; Romero-Brey, I.; Radujkovic, D.; Klein, R.; Ruggieri, A.; Rebhan, I.; Bartenschlager, R.; Lohmann, V. The lipid kinase phosphatidylinositol-4 kinase III alpha regulates the phosphorylation status of hepatitis C virus NS5A. PLoS Pathog. 2013, 9, e1003359. [Google Scholar] [CrossRef]
- Salloum, S.; Wang, H.; Ferguson, C.; Parton, R.G.; Tai, A.W. Rab18 binds to hepatitis C virus NS5A and promotes interaction between sites of viral replication and lipid droplets. PLoS Pathog. 2013, 9, e1003513. [Google Scholar] [CrossRef] [PubMed]
- Camus, G.; Herker, E.; Modi, A.A.; Haas, J.T.; Ramage, H.R.; Farese, R.V., Jr.; Ott, M. Diacylglycerol acyltransferase-1 localizes hepatitis C virus NS5A protein to lipid droplets and enhances NS5A interaction with the viral capsid core. J. Biol. Chem. 2013, 288, 9915–9923. [Google Scholar] [CrossRef]
- Nguyen, L.P.; Nguyen, T.T.T.; Nguyen, H.C.; Pham, H.T.; Han, K.M.; Choi, D.H.; Park, E.M.; Kang, S.M.; Tark, D.; Lim, Y.S.; et al. Cortactin Interacts with Hepatitis C Virus Core and NS5A Proteins: Implications for Virion Assembly. J. Virol. 2020, 94, e01306-20. [Google Scholar] [CrossRef]
- Kohler, J.J.; Nettles, J.H.; Amblard, F.; Hurwitz, S.J.; Bassit, L.; Stanton, R.A.; Ehteshami, M.; Schinazi, R.F. Approaches to hepatitis C treatment and cure using NS5A inhibitors. Infect. Drug Resist. 2014, 7, 41–56. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pawlotsky, J.M. NS5A inhibitors in the treatment of hepatitis C. J. Hepatol. 2013, 59, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Asselah, T. NS5A inhibitors: A new breakthrough for the treatment of chronic hepatitis C. J. Hepatol. 2011, 54, 1069–1072. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Zeng, Y.; Xu, S.; Wei, Y.; Zhang, X.; Wang, Q.; Jia, Y.; Wang, W.; Han, L.; Chen, Z.; Wang, Z.; et al. The PB1 protein of influenza A virus inhibits the innate immune response by targeting MAVS for NBR1-mediated selective autophagic degradation. PLoS Pathog. 2021, 17, e1009300. [Google Scholar] [CrossRef]
- Jewell, J.L.; Russell, R.C.; Guan, K.L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef]
- Gomez-Sanchez, R.; Tooze, S.A.; Reggiori, F. Membrane supply and remodeling during autophagosome biogenesis. Curr. Opin. Cell Biol. 2021, 71, 112–119. [Google Scholar] [CrossRef]
- Melia, T.J.; Lystad, A.H.; Simonsen, A. Autophagosome biogenesis: From membrane growth to closure. J. Cell Biol. 2020, 219, e202002085. [Google Scholar] [CrossRef]
- Matsunaga, K.; Morita, E.; Saitoh, T.; Akira, S.; Ktistakis, N.T.; Izumi, T.; Noda, T.; Yoshimori, T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J. Cell Biol. 2010, 190, 511–521. [Google Scholar] [CrossRef]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbe, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef]
- Yamamoto, H.; Kakuta, S.; Watanabe, T.M.; Kitamura, A.; Sekito, T.; Kondo-Kakuta, C.; Ichikawa, R.; Kinjo, M.; Ohsumi, Y. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J. Cell Biol. 2012, 198, 219–233. [Google Scholar] [CrossRef]
- Orsi, A.; Razi, M.; Dooley, H.C.; Robinson, D.; Weston, A.E.; Collinson, L.M.; Tooze, S.A. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 2012, 23, 1860–1873. [Google Scholar] [CrossRef]
- Mari, M.; Griffith, J.; Rieter, E.; Krishnappa, L.; Klionsky, D.J.; Reggiori, F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J. Cell Biol. 2010, 190, 1005–1022. [Google Scholar] [CrossRef]
- Feng, Y.; Backues, S.K.; Baba, M.; Heo, J.M.; Harper, J.W.; Klionsky, D.J. Phosphorylation of Atg9 regulates movement to the phagophore assembly site and the rate of autophagosome formation. Autophagy 2016, 12, 648–658. [Google Scholar] [CrossRef]
- Mailler, E.; Guardia, C.M.; Bai, X.; Jarnik, M.; Williamson, C.D.; Li, Y.; Maio, N.; Golden, A.; Bonifacino, J.S. The autophagy protein ATG9A enables lipid mobilization from lipid droplets. Nat. Commun. 2021, 12, 6750. [Google Scholar] [CrossRef] [PubMed]
- Molejon, M.I.; Ropolo, A.; Re, A.L.; Boggio, V.; Vaccaro, M.I. The VMP1-Beclin 1 interaction regulates autophagy induction. Sci. Rep. 2013, 3, 1055. [Google Scholar] [CrossRef] [PubMed]
- Ropolo, A.; Grasso, D.; Pardo, R.; Sacchetti, M.L.; Archange, C.; Lo Re, A.; Seux, M.; Nowak, J.; Gonzalez, C.D.; Iovanna, J.L.; et al. The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J. Biol. Chem. 2007, 282, 37124–37133. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.G.; Chen, Y.; Miao, G.; Zhao, H.; Qu, W.; Li, D.; Wang, Z.; Liu, N.; Li, L.; Chen, S.; et al. The ER-Localized Transmembrane Protein EPG-3/VMP1 Regulates SERCA Activity to Control ER-Isolation Membrane Contacts for Autophagosome Formation. Mol. Cell 2017, 67, 974–989.E6. [Google Scholar] [CrossRef]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef]
- Yla-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Shintani, T.; Nair, U.; Klionsky, D.J. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy 2005, 1, 101–109. [Google Scholar] [CrossRef]
- Yen, W.L.; Shintani, T.; Nair, U.; Cao, Y.; Richardson, B.C.; Li, Z.; Hughson, F.M.; Baba, M.; Klionsky, D.J. The conserved oligomeric Golgi complex is involved in double-membrane vesicle formation during autophagy. J. Cell Biol. 2010, 188, 101–114. [Google Scholar] [CrossRef]
- Puri, C.; Vicinanza, M.; Ashkenazi, A.; Gratian, M.J.; Zhang, Q.; Bento, C.F.; Renna, M.; Menzies, F.M.; Rubinsztein, D.C. The RAB11A-Positive Compartment Is a Primary Platform for Autophagosome Assembly Mediated by WIPI2 Recognition of PI3P-RAB11A. Dev. Cell 2018, 45, 114–131.E8. [Google Scholar] [CrossRef]
- Knaevelsrud, H.; Soreng, K.; Raiborg, C.; Haberg, K.; Rasmuson, F.; Brech, A.; Liestol, K.; Rusten, T.E.; Stenmark, H.; Neufeld, T.P.; et al. Membrane remodeling by the PX-BAR protein SNX18 promotes autophagosome formation. J. Cell Biol. 2013, 202, 331–349. [Google Scholar] [CrossRef]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Sugita, H.; Yoshimori, T.; Ohsumi, Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 1998, 273, 33889–33892. [Google Scholar] [CrossRef]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef]
- Suzuki, K.; Kirisako, T.; Kamada, Y.; Mizushima, N.; Noda, T.; Ohsumi, Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001, 20, 5971–5981. [Google Scholar] [CrossRef]
- Nakatogawa, H.; Ichimura, Y.; Ohsumi, Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007, 130, 165–178. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Codogno, P.; Zhang, H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat. Rev. Mol. Cell Biol. 2021, 22, 733–750. [Google Scholar] [CrossRef]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Noda, T.; Ohsumi, Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J. 1999, 18, 3888–3896. [Google Scholar] [CrossRef]
- Kuma, A.; Mizushima, N.; Ishihara, N.; Ohsumi, Y. Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J. Biol. Chem. 2002, 277, 18619–18625. [Google Scholar] [CrossRef]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Nair, U.; Klionsky, D.J. Atg8 controls phagophore expansion during autophagosome formation. Mol. Biol. Cell 2008, 19, 3290–3298. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Leidal, A.M.; Debnath, J.; Hansen, M. Beyond Autophagy: The Expanding Roles of ATG8 Proteins. Trends Biochem. Sci. 2021, 46, 673–686. [Google Scholar] [CrossRef]
- Kriegenburg, F.; Ungermann, C.; Reggiori, F. Coordination of Autophagosome-Lysosome Fusion by Atg8 Family Members. Curr. Biol. 2018, 28, R512–R518. [Google Scholar] [CrossRef]
- Yu, S.; Melia, T.J. The coordination of membrane fission and fusion at the end of autophagosome maturation. Curr. Opin. Cell Biol. 2017, 47, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Ungermann, C. Autophagosome Maturation and Fusion. J. Mol. Biol. 2017, 429, 486–496. [Google Scholar] [CrossRef]
- Lorincz, P.; Juhasz, G. Autophagosome-Lysosome Fusion. J. Mol. Biol. 2020, 432, 2462–2482. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef]
- Saleeb, R.S.; Kavanagh, D.M.; Dun, A.R.; Dalgarno, P.A.; Duncan, R.R. A VPS33A-binding motif on syntaxin 17 controls autophagy completion in mammalian cells. J. Biol. Chem. 2019, 294, 4188–4201. [Google Scholar] [CrossRef]
- Matsui, T.; Jiang, P.; Nakano, S.; Sakamaki, Y.; Yamamoto, H.; Mizushima, N. Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J. Cell Biol. 2018, 217, 2633–2645. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Reggiori, F.; Ungermann, C. A novel in vitro assay reveals SNARE topology and the role of Ykt6 in autophagosome fusion with vacuoles. J. Cell Biol. 2018, 217, 3670–3682. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Fan, W.; Lu, Y.; Ding, X.; Chen, S.; Zhong, Q. A mammalian autophagosome maturation mechanism mediated by TECPR1 and the Atg12-Atg5 conjugate. Mol. Cell 2012, 45, 629–641. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, L.; Lak, B.; Li, J.; Jokitalo, E.; Wang, Y. GRASP55 Senses Glucose Deprivation through O-GlcNAcylation to Promote Autophagosome-Lysosome Fusion. Dev. Cell 2018, 45, 245–261.E6. [Google Scholar] [CrossRef]
- McEwan, D.G.; Popovic, D.; Gubas, A.; Terawaki, S.; Suzuki, H.; Stadel, D.; Coxon, F.P.; Miranda de Stegmann, D.; Bhogaraju, S.; Maddi, K.; et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol. Cell 2015, 57, 39–54. [Google Scholar] [CrossRef]
- Wang, Z.; Miao, G.; Xue, X.; Guo, X.; Yuan, C.; Wang, Z.; Zhang, G.; Chen, Y.; Feng, D.; Hu, J.; et al. The Vici Syndrome Protein EPG5 Is a Rab7 Effector that Determines the Fusion Specificity of Autophagosomes with Late Endosomes/Lysosomes. Mol. Cell 2016, 63, 781–795. [Google Scholar] [CrossRef]
- Ebner, P.; Poetsch, I.; Deszcz, L.; Hoffmann, T.; Zuber, J.; Ikeda, F. The IAP family member BRUCE regulates autophagosome-lysosome fusion. Nat. Commun. 2018, 9, 599. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Munafo, D.B.; Beron, W.; Colombo, M.I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004, 117, 2687–2697. [Google Scholar] [CrossRef]
- Pankiv, S.; Johansen, T. FYCO1: Linking autophagosomes to microtubule plus end-directing molecular motors. Autophagy 2010, 6, 550–552. [Google Scholar] [CrossRef]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.A.; Lamark, T.; Overvatn, A.; Bjorkoy, G.; Johansen, T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Jordens, I.; Fernandez-Borja, M.; Marsman, M.; Dusseljee, S.; Janssen, L.; Calafat, J.; Janssen, H.; Wubbolts, R.; Neefjes, J. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol. 2001, 11, 1680–1685. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Cantalupo, G.; Alifano, P.; Roberti, V.; Bruni, C.B.; Bucci, C. Rab-interacting lysosomal protein (RILP): The Rab7 effector required for transport to lysosomes. EMBO J. 2001, 20, 683–693. [Google Scholar] [CrossRef]
- Khobrekar, N.V.; Quintremil, S.; Dantas, T.J.; Vallee, R.B. The Dynein Adaptor RILP Controls Neuronal Autophagosome Biogenesis, Transport, and Clearance. Dev. Cell 2020, 53, 141–153.E4. [Google Scholar] [CrossRef]
- Wijdeven, R.H.; Janssen, H.; Nahidiazar, L.; Janssen, L.; Jalink, K.; Berlin, I.; Neefjes, J. Cholesterol and ORP1L-mediated ER contact sites control autophagosome transport and fusion with the endocytic pathway. Nat. Commun. 2016, 7, 11808. [Google Scholar] [CrossRef]
- Johansson, M.; Rocha, N.; Zwart, W.; Jordens, I.; Janssen, L.; Kuijl, C.; Olkkonen, V.M.; Neefjes, J. Activation of endosomal dynein motors by stepwise assembly of Rab7-RILP-p150Glued, ORP1L, and the receptor betalll spectrin. J. Cell Biol. 2007, 176, 459–471. [Google Scholar] [CrossRef]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, F.; Jian, F.; Rong, Y. Recent progresses in the late stages of autophagy. Cell Insight 2024, 3, 100152. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Z.; Du, W.; Que, H.; Wang, Y.; Ouyang, Q.; Jian, F.; Yuan, W.; Zhao, Y.; Tian, R.; et al. Recycling of autophagosomal components from autolysosomes by the recycler complex. Nat. Cell Biol. 2022, 24, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhou, C.; Que, H.; Wang, Y.; Rong, Y. The fate of autophagosomal membrane components. Autophagy 2023, 19, 370–371. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Zaffagnini, G.; Martens, S. Mechanisms of Selective Autophagy. J. Mol. Biol. 2016, 428, 1714–1724. [Google Scholar] [CrossRef]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; Rogov, V.V. A Diversity of Selective Autophagy Receptors Determines the Specificity of the Autophagy Pathway. Mol. Cell 2019, 76, 268–285. [Google Scholar] [CrossRef]
- Vargas, J.N.S.; Hamasaki, M.; Kawabata, T.; Youle, R.J.; Yoshimori, T. The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol. 2023, 24, 167–185. [Google Scholar] [CrossRef]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef]
- Birgisdottir, A.B.; Lamark, T.; Johansen, T. The LIR motif - crucial for selective autophagy. J. Cell Sci. 2013, 126, 3237–3247. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Lamark, T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Biol. 2020, 432, 80–103. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef]
- Adriaenssens, E.; Ferrari, L.; Martens, S. Orchestration of selective autophagy by cargo receptors. Curr. Biol. 2022, 32, R1357–R1371. [Google Scholar] [CrossRef]
- Rogov, V.V.; Stolz, A.; Ravichandran, A.C.; Rios-Szwed, D.O.; Suzuki, H.; Kniss, A.; Lohr, F.; Wakatsuki, S.; Dotsch, V.; Dikic, I.; et al. Structural and functional analysis of the GABARAP interaction motif (GIM). EMBO Rep. 2017, 18, 1382–1396. [Google Scholar] [CrossRef] [PubMed]
- Fracchiolla, D.; Sawa-Makarska, J.; Martens, S. Beyond Atg8 binding: The role of AIM/LIR motifs in autophagy. Autophagy 2017, 13, 978–979. [Google Scholar] [CrossRef]
- Schaaf, M.B.; Keulers, T.G.; Vooijs, M.A.; Rouschop, K.M. LC3/GABARAP family proteins: Autophagy-(un)related functions. FASEB J. 2016, 30, 3961–3978. [Google Scholar] [CrossRef] [PubMed]
- Jacomin, A.C.; Samavedam, S.; Promponas, V.; Nezis, I.P. iLIR database: A web resource for LIR motif-containing proteins in eukaryotes. Autophagy 2016, 12, 1945–1953. [Google Scholar] [CrossRef]
- Okamoto, K. Organellophagy: Eliminating cellular building blocks via selective autophagy. J. Cell Biol. 2014, 205, 435–445. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Uoselis, L.; Nguyen, T.N.; Lazarou, M. Mitochondrial degradation: Mitophagy and beyond. Mol. Cell 2023, 83, 3404–3420. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef]
- Meissner, C.; Lorenz, H.; Weihofen, A.; Selkoe, D.J.; Lemberg, M.K. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 2011, 117, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Yang, J.Y.; Yang, W.Y. Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat. Commun. 2013, 4, 2428. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Hoshino, A.; Wang, W.J.; Wada, S.; McDermott-Roe, C.; Evans, C.S.; Gosis, B.; Morley, M.P.; Rathi, K.S.; Li, J.; Li, K.; et al. The ADP/ATP translocase drives mitophagy independent of nucleotide exchange. Nature 2019, 575, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Psakhye, I.; Jentsch, S. Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell 2014, 158, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.A.; Phillips, E.O.; Collier, C.L.; Jb Robinson, A.; Routledge, D.; Wood, R.E.; Assar, E.A.; Tumbarello, D.A. Tollip coordinates Parkin-dependent trafficking of mitochondrial-derived vesicles. EMBO J. 2020, 39, e102539. [Google Scholar] [CrossRef] [PubMed]
- Di Rienzo, M.; Romagnoli, A.; Ciccosanti, F.; Refolo, G.; Consalvi, V.; Arena, G.; Valente, E.M.; Piacentini, M.; Fimia, G.M. AMBRA1 regulates mitophagy by interacting with ATAD3A and promoting PINK1 stability. Autophagy 2022, 18, 1752–1762. [Google Scholar] [CrossRef]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 517. [Google Scholar] [CrossRef]
- Van Humbeeck, C.; Cornelissen, T.; Hofkens, H.; Mandemakers, W.; Gevaert, K.; De Strooper, B.; Vandenberghe, W. Parkin interacts with Ambra1 to induce mitophagy. J. Neurosci. 2011, 31, 10249–10261. [Google Scholar] [CrossRef]
- Ding, W.X.; Ni, H.M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W., 2nd; Yin, X.M. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Lohr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef]
- Quinsay, M.N.; Thomas, R.L.; Lee, Y.; Gustafsson, A.B. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 2010, 6, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238.E10. [Google Scholar] [CrossRef]
- Yan, C.; Gong, L.; Chen, L.; Xu, M.; Abou-Hamdan, H.; Tang, M.; Desaubry, L.; Song, Z. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy 2020, 16, 419–434. [Google Scholar] [CrossRef]
- Sun, S.; Hou, H.; Ma, G.; Ma, Q.; Li, N.; Zhang, L.; Dong, C.; Cao, M.; Tam, K.Y.; Ying, Z.; et al. The interaction between E3 ubiquitin ligase Parkin and mitophagy receptor PHB2 links inner mitochondrial membrane ubiquitination to efficient mitophagy. J. Biol. Chem. 2022, 298, 102704. [Google Scholar] [CrossRef]
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef]
- Matsumoto, G.; Shimogori, T.; Hattori, N.; Nukina, N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet. 2015, 24, 4429–4442. [Google Scholar] [CrossRef]
- Hsieh, C.W.; Yang, W.Y. Omegasome-proximal PtdIns(4,5)P2 couples F-actin mediated mitoaggregate disassembly with autophagosome formation during mitophagy. Nat. Commun. 2019, 10, 969. [Google Scholar] [CrossRef]
- Ait-Goughoulte, M.; Kanda, T.; Meyer, K.; Ryerse, J.S.; Ray, R.B.; Ray, R. Hepatitis C virus genotype 1a growth and induction of autophagy. J. Virol. 2008, 82, 2241–2249. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 2005, 102, 9294–9299. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef]
- Ke, P.Y.; Chen, S.S. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Investig. 2011, 121, 37–56. [Google Scholar] [CrossRef]
- Shrivastava, S.; Raychoudhuri, A.; Steele, R.; Ray, R.; Ray, R.B. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 2011, 53, 406–414. [Google Scholar] [CrossRef]
- Chan, S.T.; Lee, J.; Narula, M.; Ou, J.J. Suppression of Host Innate Immune Response by Hepatitis C Virus via Induction of Autophagic Degradation of TRAF6. J. Virol. 2016, 90, 10928–10935. [Google Scholar] [CrossRef]
- Chandra, P.K.; Bao, L.; Song, K.; Aboulnasr, F.M.; Baker, D.P.; Shores, N.; Wimley, W.C.; Liu, S.; Hagedorn, C.H.; Fuchs, S.Y.; et al. HCV infection selectively impairs type I but not type III IFN signaling. Am. J. Pathol. 2014, 184, 214–229. [Google Scholar] [CrossRef]
- Mori, H.; Fukuhara, T.; Ono, C.; Tamura, T.; Sato, A.; Fauzyah, Y.; Wada, M.; Okamoto, T.; Noda, T.; Yoshimori, T.; et al. Induction of selective autophagy in cells replicating hepatitis C virus genome. J. Gen. Virol. 2018, 99, 1643–1657. [Google Scholar] [CrossRef] [PubMed]
- Vescovo, T.; Romagnoli, A.; Perdomo, A.B.; Corazzari, M.; Ciccosanti, F.; Alonzi, T.; Nardacci, R.; Ippolito, G.; Tripodi, M.; Garcia-Monzon, C.; et al. Autophagy protects cells from HCV-induced defects in lipid metabolism. Gastroenterology 2012, 142, 644–653.e3. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Syed, G.H.; Siddiqui, A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 2013, 9, e1003285. [Google Scholar] [CrossRef]
- Kim, N.; Kim, M.J.; Sung, P.S.; Bae, Y.C.; Shin, E.C.; Yoo, J.Y. Interferon-inducible protein SCOTIN interferes with HCV replication through the autolysosomal degradation of NS5A. Nat. Commun. 2016, 7, 10631. [Google Scholar] [CrossRef]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef]
- Gkikas, I.; Palikaras, K.; Tavernarakis, N. The Role of Mitophagy in Innate Immunity. Front. Immunol. 2018, 9, 1283. [Google Scholar] [CrossRef]
- Fu, C.; Cao, N.; Liu, W.; Zhang, Z.; Yang, Z.; Zhu, W.; Fan, S. Crosstalk between mitophagy and innate immunity in viral infection. Front. Microbiol. 2022, 13, 1064045. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.H.; Kim, J.K.; Jo, E.K. Mitophagy and Innate Immunity in Infection. Mol. Cells 2020, 43, 10–22. [Google Scholar] [CrossRef]
- Lazarou, M. Keeping the immune system in check: A role for mitophagy. Immunol. Cell Biol. 2015, 93, 3–10. [Google Scholar] [CrossRef]
- Moehlman, A.T.; Youle, R.J. Mitochondrial Quality Control and Restraining Innate Immunity. Annu. Rev. Cell Dev. Biol. 2020, 36, 265–289. [Google Scholar] [CrossRef]
- Negash, A.A.; Ramos, H.J.; Crochet, N.; Lau, D.T.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; Hagedorn, C.H.; et al. IL-1beta production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013, 9, e1003330. [Google Scholar] [CrossRef] [PubMed]
- Negash, A.A.; Olson, R.M.; Griffin, S.; Gale, M., Jr. Modulation of calcium signaling pathway by hepatitis C virus core protein stimulates NLRP3 inflammasome activation. PLoS Pathog. 2019, 15, e1007593. [Google Scholar] [CrossRef]
- Daussy, C.F.; Monard, S.C.; Guy, C.; Munoz-Gonzalez, S.; Chazal, M.; Anthonsen, M.W.; Jouvenet, N.; Henry, T.; Dreux, M.; Meurs, E.F.; et al. The Inflammasome Components NLRP3 and ASC Act in Concert with IRGM To Rearrange the Golgi Apparatus during Hepatitis C Virus Infection. J. Virol. 2021, 95, e00826-20. [Google Scholar] [CrossRef]
- Gupta, S.; Cassel, S.L.; Sutterwala, F.S.; Dagvadorj, J. Regulation of the NLRP3 inflammasome by autophagy and mitophagy. Immunol. Rev. 2024. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.M.; Silwal, P.; Jo, E.K. Inflammasome and Mitophagy Connection in Health and Disease. Int. J. Mol. Sci. 2020, 21, 4714. [Google Scholar] [CrossRef]
- Mohl, B.P.; Bartlett, C.; Mankouri, J.; Harris, M. Early events in the generation of autophagosomes are required for the formation of membrane structures involved in hepatitis C virus genome replication. J. Gen. Virol. 2016, 97, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kim, J.Y.; Liu, H.M.; Lai, M.M.C.; Ou, J.J. HCV-induced autophagosomes are generated via homotypic fusion of phagophores that mediate HCV RNA replication. PLoS Pathog. 2017, 13, e1006609. [Google Scholar] [CrossRef]
- Wang, L.; Tian, Y.; Ou, J.H. HCV induces the expression of Rubicon and UVRAG to temporally regulate the maturation of autophagosomes and viral replication. PLoS Pathog. 2015, 11, e1004764. [Google Scholar] [CrossRef]
- Sir, D.; Kuo, C.F.; Tian, Y.; Liu, H.M.; Huang, E.J.; Jung, J.U.; Machida, K.; Ou, J.H. Replication of hepatitis C virus RNA on autophagosomal membranes. J. Biol. Chem. 2012, 287, 18036–18043. [Google Scholar] [CrossRef]
- Guevin, C.; Manna, D.; Belanger, C.; Konan, K.V.; Mak, P.; Labonte, P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 2010, 405, 1–7. [Google Scholar] [CrossRef]
- Tanida, I.; Fukasawa, M.; Ueno, T.; Kominami, E.; Wakita, T.; Hanada, K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009, 5, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Devhare, P.; Sujijantarat, N.; Steele, R.; Kwon, Y.C.; Ray, R.; Ray, R.B. Knockdown of Autophagy Inhibits Infectious Hepatitis C Virus Release by the Exosomal Pathway. J. Virol. 2016, 90, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Ou, J.J. Regulation of Apolipoprotein E Trafficking by Hepatitis C Virus-induced Autophagy. J. Virol. 2018, 92, e00211-18. [Google Scholar] [CrossRef]
- Su, W.C.; Chao, T.C.; Huang, Y.L.; Weng, S.C.; Jeng, K.S.; Lai, M.M. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J. Virol. 2011, 85, 10561–10571. [Google Scholar] [CrossRef]
- Gregoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 2011, 7, e1002422. [Google Scholar] [CrossRef]
- Wang, J.; Kang, R.; Huang, H.; Xi, X.; Wang, B.; Wang, J.; Zhao, Z. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy 2014, 10, 766–784. [Google Scholar] [CrossRef]
- Hara, Y.; Yanatori, I.; Ikeda, M.; Kiyokage, E.; Nishina, S.; Tomiyama, Y.; Toida, K.; Kishi, F.; Kato, N.; Imamura, M.; et al. Hepatitis C virus core protein suppresses mitophagy by interacting with parkin in the context of mitochondrial depolarization. Am. J. Pathol. 2014, 184, 3026–3039. [Google Scholar] [CrossRef] [PubMed]
- Quan, M.; Liu, S.; Li, G.; Wang, Q.; Zhang, J.; Zhang, M.; Li, M.; Gao, P.; Feng, S.; Cheng, J. A functional role for NS5ATP9 in the induction of HCV NS5A-mediated autophagy. J. Viral Hepat. 2014, 21, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Siu, G.K.; Zhou, F.; Yu, M.K.; Zhang, L.; Wang, T.; Liang, Y.; Chen, Y.; Chan, H.C.; Yu, S. Hepatitis C virus NS5A protein cooperates with phosphatidylinositol 4-kinase IIIalpha to induce mitochondrial fragmentation. Sci. Rep. 2016, 6, 23464. [Google Scholar] [CrossRef]
- Ke, P.Y. Mitophagy in the Pathogenesis of Liver Diseases. Cells 2020, 9, 831. [Google Scholar] [CrossRef]
- Zhu, L.; Wu, X.; Liao, R. Mechanism and regulation of mitophagy in nonalcoholic fatty liver disease (NAFLD): A mini-review. Life Sci. 2023, 312, 121162. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Long, H.; Hou, L.; Feng, B.; Ma, Z.; Wu, Y.; Zeng, Y.; Cai, J.; Zhang, D.W.; Zhao, G. The mitophagy pathway and its implications in human diseases. Signal Transduct. Target. Ther. 2023, 8, 304. [Google Scholar] [CrossRef]
- Ma, X.; McKeen, T.; Zhang, J.; Ding, W.X. Role and Mechanisms of Mitophagy in Liver Diseases. Cells 2020, 9, 837. [Google Scholar] [CrossRef]
- Liu, K.; Lee, J.; Kim, J.Y.; Wang, L.; Tian, Y.; Chan, S.T.; Cho, C.; Machida, K.; Chen, D.; Ou, J.J. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol. Cell 2017, 68, 281–292.E5. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Ding, P.; Song, Y.; Zhou, J.; Chen, X.; Peng, C.; Liu, S. FDX1 downregulation activates mitophagy and the PI3K/AKT signaling pathway to promote hepatocellular carcinoma progression by inducing ROS production. Redox Biol. 2024, 75, 103302. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cheng, H.; Li, M.; Gao, D.; Wu, H.; Zhang, S.; Huang, Y.; Guo, K. BNIP3-mediated mitophagy boosts the competitive growth of Lenvatinib-resistant cells via energy metabolism reprogramming in HCC. Cell Death Dis. 2024, 15, 484. [Google Scholar] [CrossRef]
- Matsui, C.; Shoji, I.; Kaneda, S.; Sianipar, I.R.; Deng, L.; Hotta, H. Hepatitis C virus infection suppresses GLUT2 gene expression via downregulation of hepatocyte nuclear factor 1alpha. J. Virol. 2012, 86, 12903–12911. [Google Scholar] [CrossRef]
- Hechtman, J.F.; Abou-Alfa, G.K.; Stadler, Z.K.; Mandelker, D.L.; Roehrl, M.H.A.; Zehir, A.; Vakiani, E.; Middha, S.; Klimstra, D.S.; Shia, J. Somatic HNF1A mutations in the malignant transformation of hepatocellular adenomas: A retrospective analysis of data from MSK-IMPACT and TCGA. Hum. Pathol. 2019, 83, 1–6. [Google Scholar] [CrossRef]
- Wang, X.; Hassan, W.; Zhao, J.; Bakht, S.; Nie, Y.; Wang, Y.; Pang, Q.; Huang, Z. The impact of hepatocyte nuclear factor-1alpha on liver malignancies and cell stemness with metabolic consequences. Stem Cell Res. Ther. 2019, 10, 315. [Google Scholar] [CrossRef]
- Yen, C.L.; Stone, S.J.; Koliwad, S.; Harris, C.; Farese, R.V., Jr. Thematic review series: Glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 2008, 49, 2283–2301. [Google Scholar] [CrossRef]
- Herker, E.; Harris, C.; Hernandez, C.; Carpentier, A.; Kaehlcke, K.; Rosenberg, A.R.; Farese, R.V., Jr.; Ott, M. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat. Med. 2010, 16, 1295–1298. [Google Scholar] [CrossRef] [PubMed]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, H.; Iijima, H.; Gaber, E.S.; Zaitsu, T.; Matsuda, M.; Wakae, K.; Watashi, K.; Suzuki, R.; Masaki, T.; Kahn, J.; et al. Hepatocellular organellar abnormalities following elimination of hepatitis C virus. Liver Int. 2023, 43, 1677–1690. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).