

Whole-Genome Sequencing and Mutation Analyses of SARS-CoV-2 Isolates from Indonesia

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Population
2.2. Sample Collection, RNA Extraction, and SARS-CoV-2 Detection
2.3. Whole-Genome Sequencing
2.4. Data Analysis
2.5. Ethical Issues
3. Results
3.1. Demographic Data of SARS-CoV-2 Infected Patients
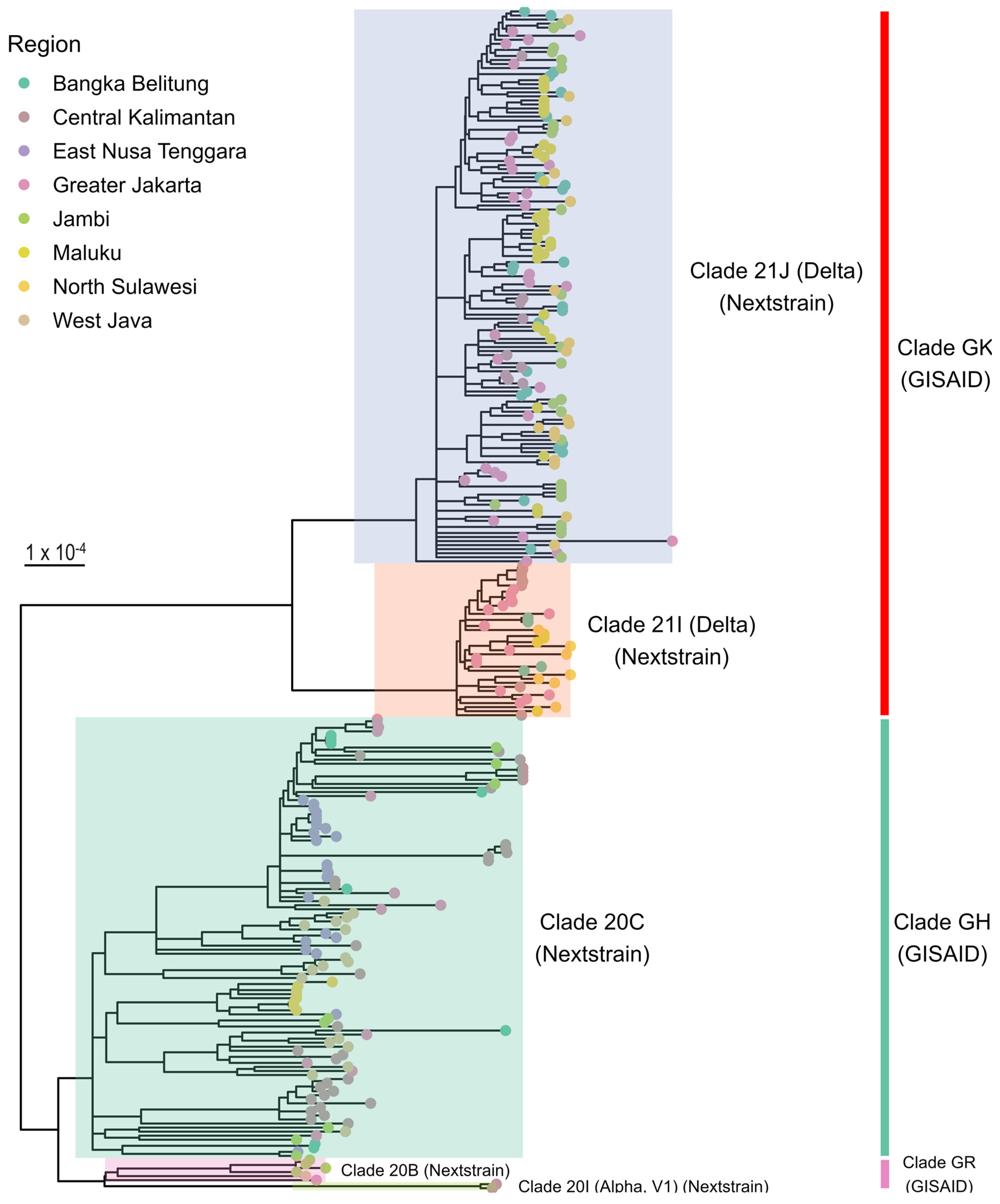
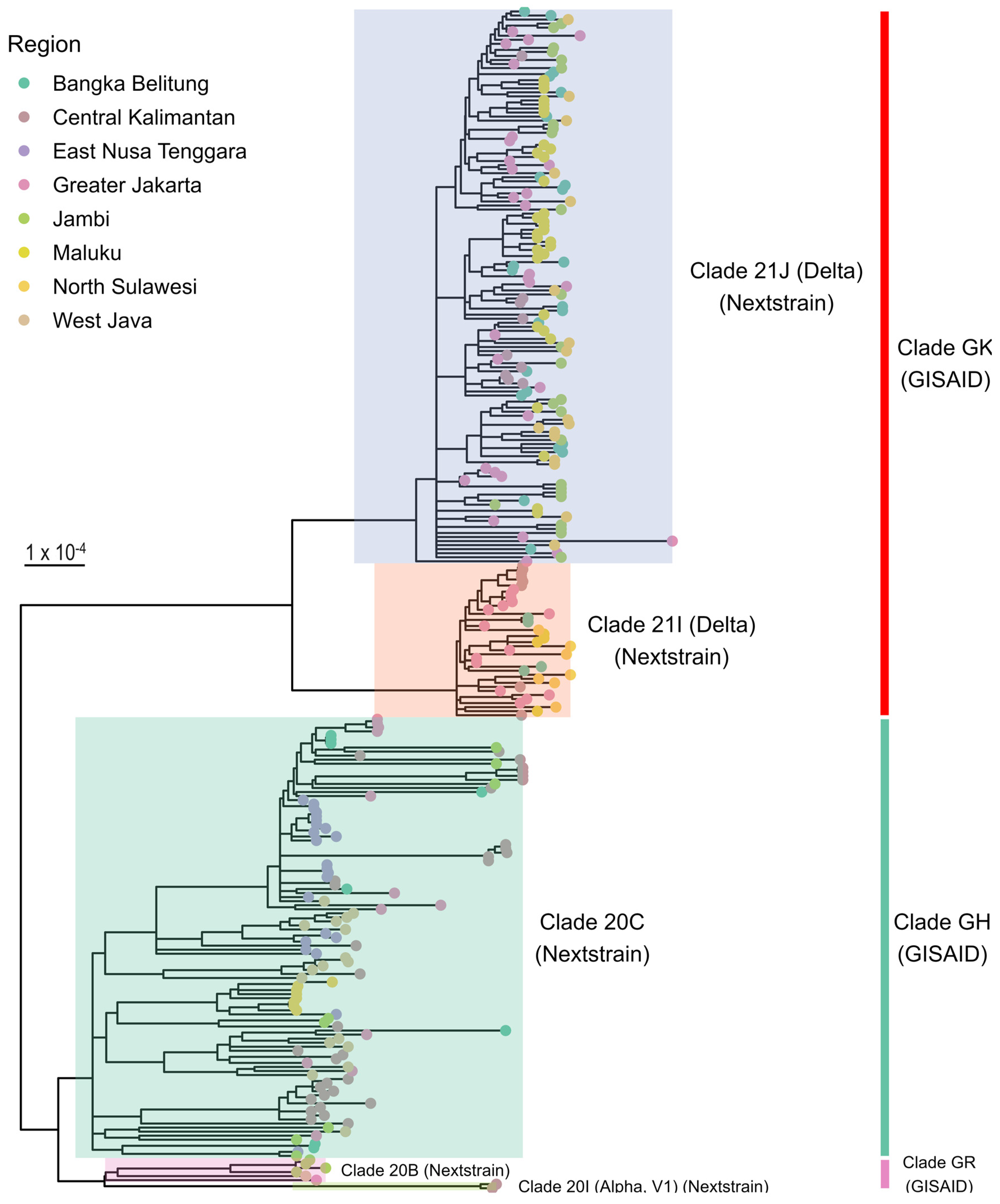
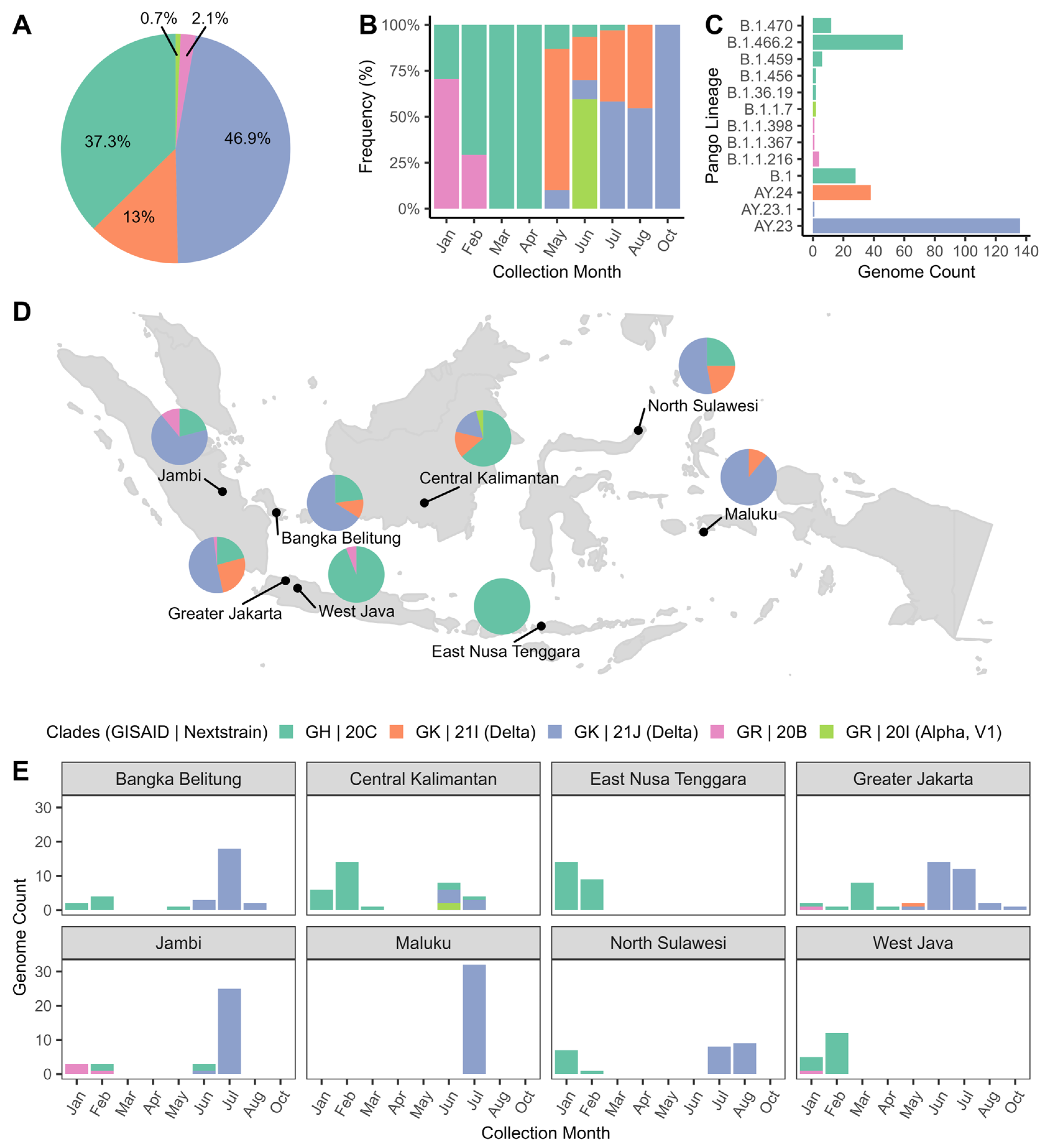
3.2. Phylogenetic Analysis
3.3. Distribution of SARS-CoV-2 Clades and Lineages
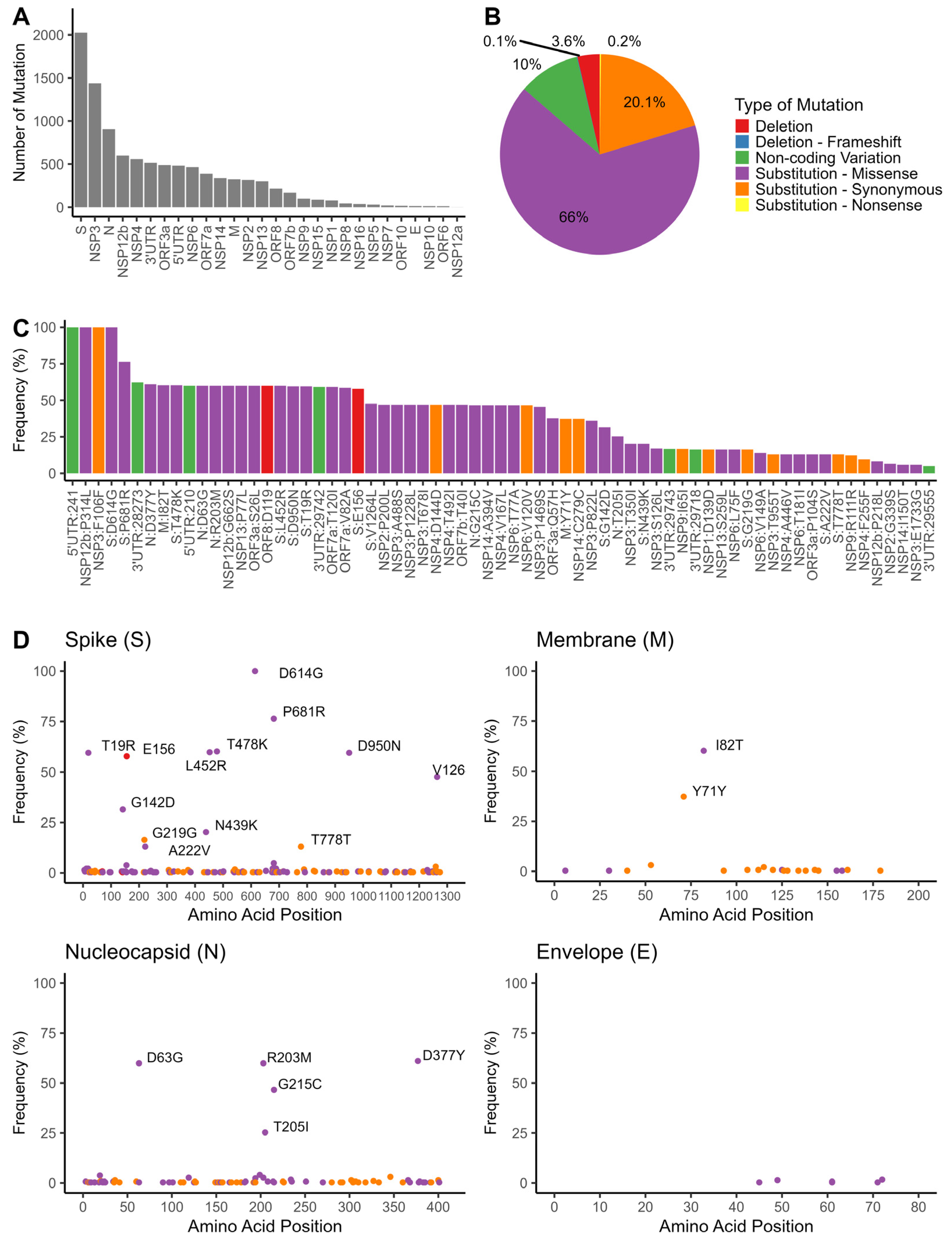
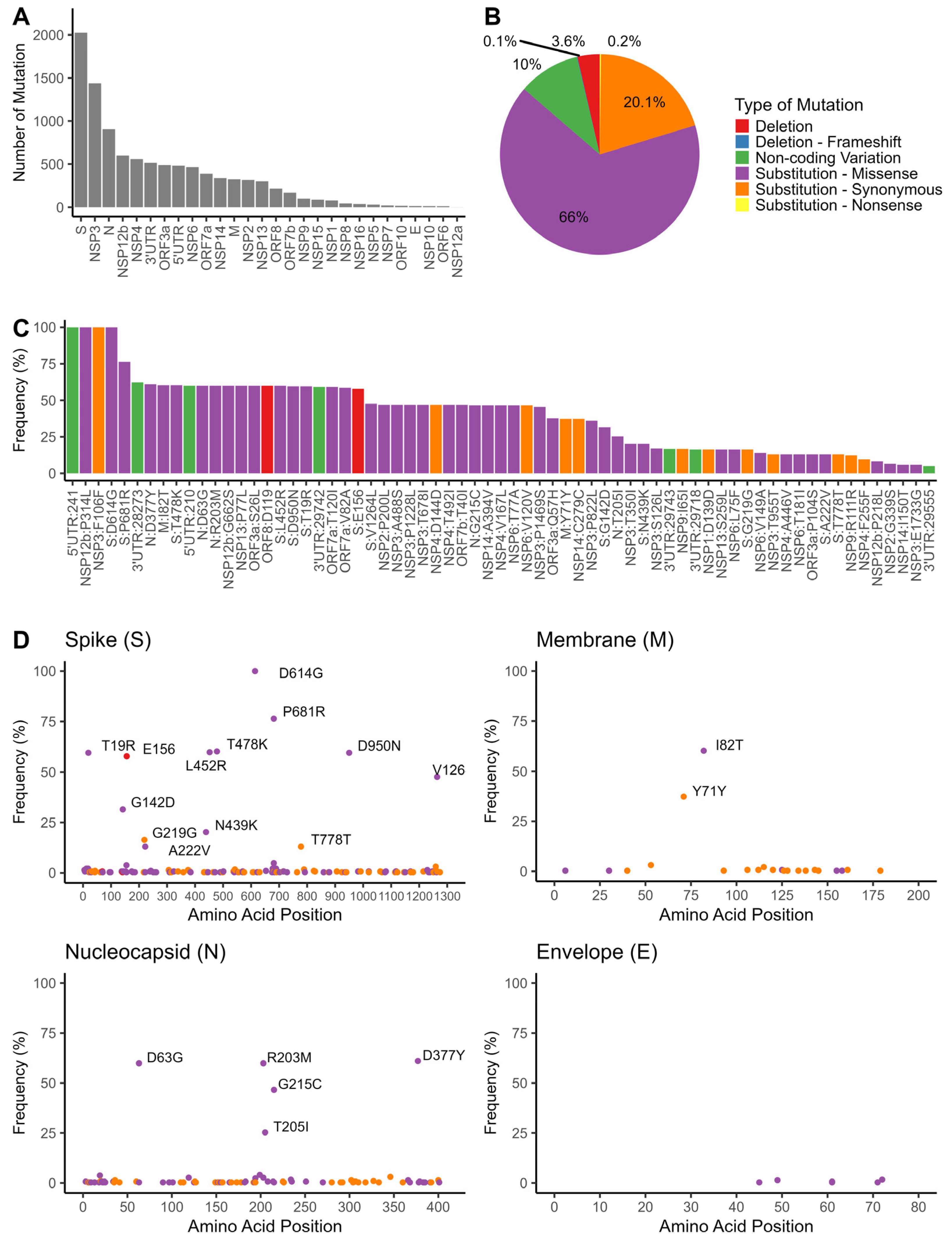
3.4. Mutation Analyses of SARS-CoV-2 Genomes
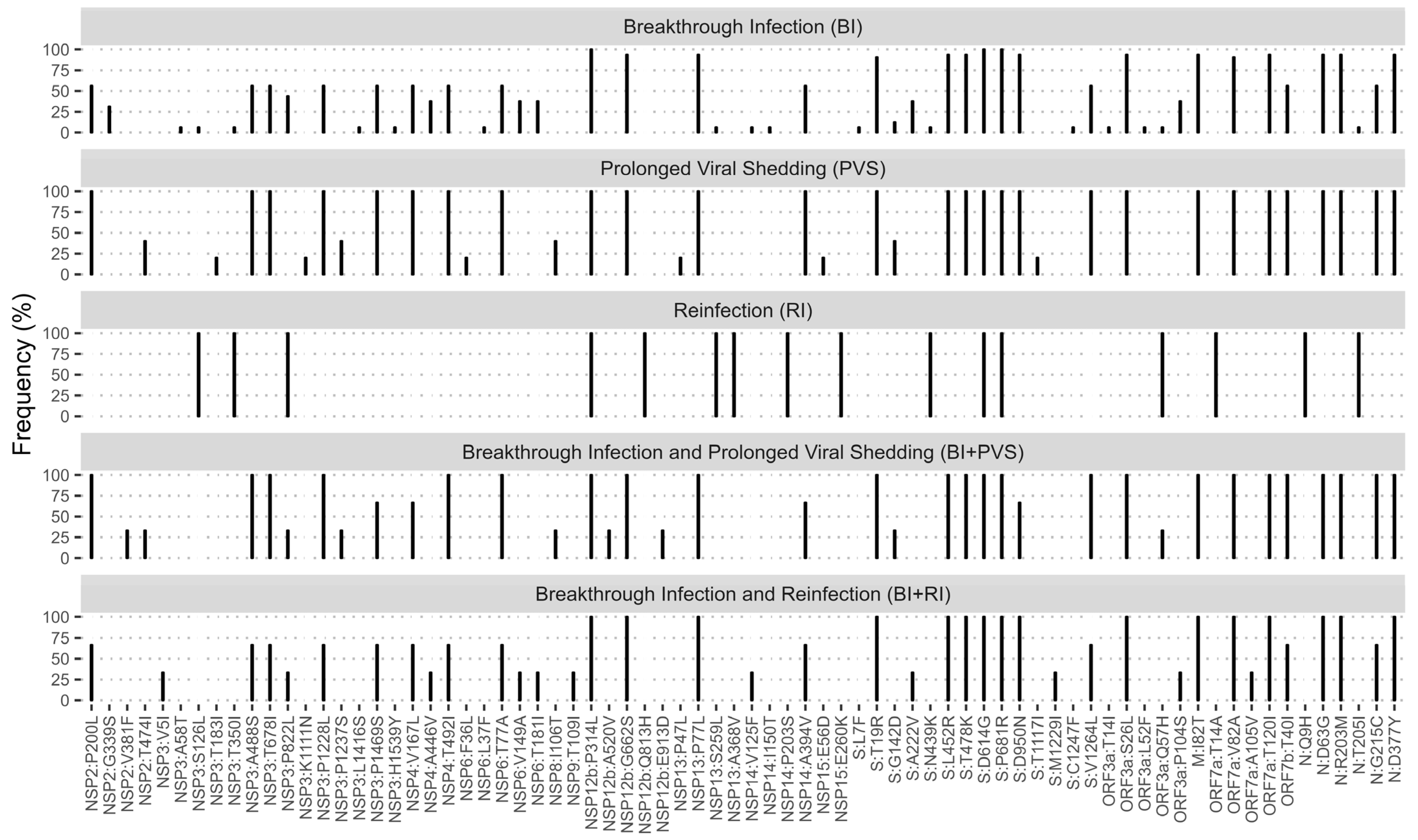
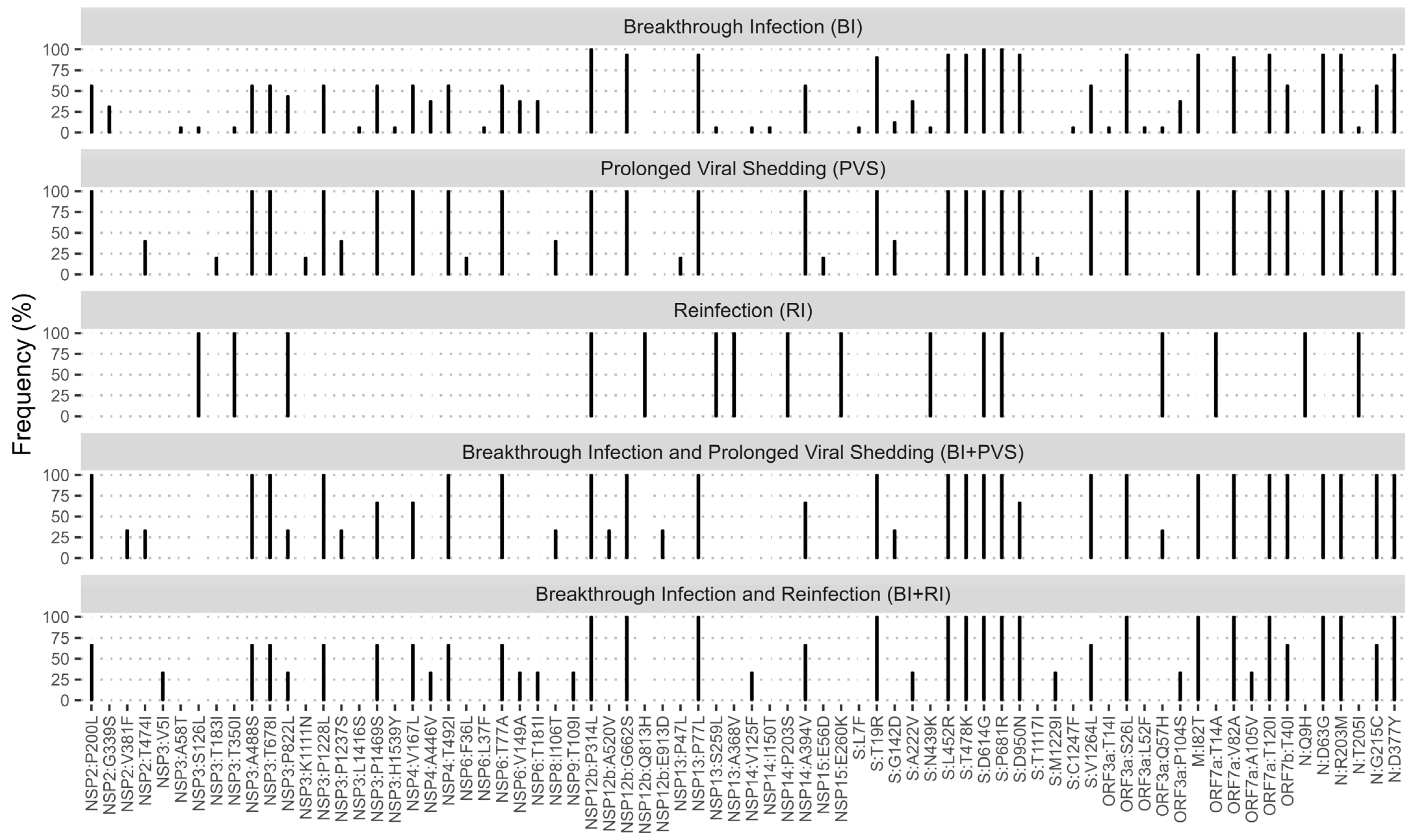
3.5. Mutation Patterns among Patients with Breakthrough Infection, Reinfection, and Prolonged Viral Shedding
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 5 October 2023).
- Duerr, R.; Dimartino, D.; Marier, C.; Zappile, P.; Levine, S.; Francois, F.; Iturrate, E.; Wang, G.; Dittmann, M.; Lighter, J.; et al. Clinical and Genomic Signatures of SARS-CoV-2 Delta Breakthrough Infections in New York. eBioMedicine 2022, 82, 104141. [Google Scholar] [CrossRef]
- Shahapur, P.R.; Shahapur, R.; Bagali, S.; Karigoudar, R.; Wavare, D.S.P.J.; Kandi, V.; Suvvari, T.K.; Mittal, R.J.; Jadhav, M. Breakthrough Infections: Clinical Profile and Outcomes of COVID-19 Vaccinated and Unvaccinated People from a Tertiary Care Hospital. Cureus 2022, 14, e32089. [Google Scholar] [CrossRef] [PubMed]
- Puhach, O.; Meyer, B.; Eckerle, I. SARS-CoV-2 Viral Load and Shedding Kinetics. Nat. Rev. Microbiol. 2023, 21, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 Genome, Structure, Evolution, Pathogenesis and Therapies: Structural Genomics Approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef] [PubMed]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; de Silva, T.I.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 Variant Biology: Immune Escape, Transmission and Fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, bbab375. [Google Scholar] [CrossRef] [PubMed]
- Tosta, S.; Moreno, K.; Schuab, G.; Fonseca, V.; Segovia, F.M.C.; Kashima, S.; Elias, M.C.; Sampaio, S.C.; Ciccozzi, M.; Alcantara, L.C.J.; et al. Global SARS-CoV-2 Genomic Surveillance: What We Have Learned (so Far). Infect. Genet. Evol. 2023, 108, 105405. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Zeng, Q.; Saputro, B.I.L.; Chew, S.P.; Chew, I.; Frendy, H.; Tan, J.W.; Li, L. Tracking the Molecular Evolution and Transmission Patterns of SARS-CoV-2 Lineage B.1.466.2 in Indonesia Based on Genomic Surveillance Data. Virol. J. 2022, 19, 103. [Google Scholar] [CrossRef]
- Cahyani, I.; Putro, E.W.; Ridwanuloh, A.M.; Wibowo, S.; Hariyatun, H.; Syahputra, G.; Akbariani, G.; Utomo, A.R.; Ilyas, M.; Loose, M.; et al. Genome Profiling of SARS-CoV-2 in Indonesia, ASEAN and the Neighbouring East Asian Countries: Features, Challenges and Achievements. Viruses 2022, 14, 778. [Google Scholar] [CrossRef]
- Massi, M.N.; Abidin, R.S.; Farouk, A.-E.; Halik, H.; Soraya, G.V.; Hidayah, N.; Sjahril, R.; Handayani, I.; Hakim, M.S.; Gazali, F.M.; et al. Full-Genome Sequencing and Mutation Analysis of SARS-CoV-2 Isolated from Makassar, South Sulawesi, Indonesia. PeerJ 2022, 10, e13522. [Google Scholar] [CrossRef]
- Fibriani, A.; Stephanie, R.; Alfiantie, A.A.; Siregar, A.L.F.; Pradani, G.A.P.; Yamahoki, N.; Purba, W.S.; Alamanda, C.N.C.; Rahmawati, E.; Rachman, R.W.; et al. Analysis of SARS-CoV-2 Genomes from West Java, Indonesia. Viruses 2021, 13, 2097. [Google Scholar] [CrossRef] [PubMed]
- Pradipta, A.; Kumaheri, M.A.; Wahyudi, L.D.; Susanto, A.P.; Agasi, H.I.; Shankar, A.H.; Sudarmono, P. Accelerating Detection of Variants During COVID-19 Surges by Diverse Technological and Public Health Partnerships: A Case Study from Indonesia. Front. Genet. 2022, 13, 801332. [Google Scholar] [CrossRef] [PubMed]
- Bhoyar, R.C.; Jain, A.; Sehgal, P.; Divakar, M.K.; Sharma, D.; Imran, M.; Jolly, B.; Ranjan, G.; Rophina, M.; Sharma, S.; et al. High Throughput Detection and Genetic Epidemiology of SARS-CoV-2 Using COVIDSeq next-Generation Sequencing. PLoS ONE 2021, 16, e0247115. [Google Scholar] [CrossRef] [PubMed]
- Trimarsanto, H. Ncov19-Pipeline. Available online: https://github.com/trmznt/ncov19-pipeline (accessed on 30 October 2023).
- Bushnell, B. BBTools. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 20 March 2023).
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An Amplicon-Based Sequencing Framework for Accurately Measuring Intrahost Virus Diversity Using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. CCDCW 2021, 3, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- GISAID Global Initiative on Sharing All Influenza Data (GISAID). Clade and Lineage Nomenclature Aids in Genomic Epidemiology Studies of Active hCoV-19 Viruses; GISAID: Munich, Germany, 2021. [Google Scholar]
- Nextstrain: Genomic Epidemiology of Novel Coronavirus—Global Subsampling. Available online: https://nextstrain.org/ncov (accessed on 25 November 2022).
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Mercatelli, D.; Triboli, L.; Fornasari, E.; Ray, F.; Giorgi, F.M. Coronapp: A Web Application to Annotate and Monitor SARS-CoV-2 Mutations. J. Med. Virol. 2021, 93, 3238–3245. [Google Scholar] [CrossRef]
- Hoan, N.X.; Pallerla, S.R.; Huy, P.X.; Krämer, H.; My, T.N.; Tung, T.T.; Hoan, P.Q.; Toan, N.L.; Song, L.H.; Velavan, T.P. SARS-CoV-2 Viral Dynamics of the First 1000 Sequences from Vietnam and Neighbouring ASEAN Countries. IJID Reg. 2022, 2, 175–179. [Google Scholar] [CrossRef]
- Fan, Y.; Li, X.; Zhang, L.; Wan, S.; Zhang, L.; Zhou, F. SARS-CoV-2 Omicron Variant: Recent Progress and Future Perspectives. Signal Transduct. Target. Ther. 2022, 7, 141. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhou, J.; Tian, M.; Huang, M.; Liu, S.; Xie, Y.; Han, P.; Bai, C.; Han, P.; Zheng, A.; et al. Omicron SARS-CoV-2 Mutations Stabilize Spike up-RBD Conformation and Lead to a Non-RBM-Binding Monoclonal Antibody Escape. Nat. Commun. 2022, 13, 4958. [Google Scholar] [CrossRef]
- Kumar, S.; Delipan, R.; Chakraborty, D.; Kanjo, K.; Singh, R.; Singh, N.; Siddiqui, S.; Tyagi, A.; Jha, V.; Thakur, K.G.; et al. Mutations in S2 Subunit of SARS-CoV-2 Omicron Spike Strongly Influence Its Conformation, Fusogenicity, and Neutralization Sensitivity. J. Virol. 2023, 97, e00922-23. [Google Scholar] [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, A.; Toptan, T.; Pallas, C.; Wolf, T.; Goetsch, U.; Gottschalk, R.; Vehreschild, M.J.G.T.; Ciesek, S.; Widera, M. Antibody-Mediated Neutralization of Authentic SARS-CoV-2 B.1.617 Variants Harboring L452R and T478K/E484Q. Viruses 2021, 13, 1693. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef] [PubMed]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 Spike-Protein D614G Mutation Increases Virion Spike Density and Infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta Spike P681R Mutation Enhances SARS-CoV-2 Fitness over Alpha Variant. Cell Rep. 2022, 39, 110829. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced Sensitivity of SARS-CoV-2 Variant Delta to Antibody Neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Mishra, T.; Dalavi, R.; Joshi, G.; Kumar, A.; Pandey, P.; Shukla, S.; Mishra, R.K.; Chande, A. SARS-CoV-2 Spike E156G/Δ157-158 Mutations Contribute to Increased Infectivity and Immune Escape. Life Sci. Alliance 2022, 5, e202201415. [Google Scholar] [CrossRef] [PubMed]
- Ginex, T.; Marco-Marín, C.; Wieczór, M.; Mata, C.P.; Krieger, J.; Ruiz-Rodriguez, P.; López-Redondo, M.L.; Francés-Gómez, C.; Melero, R.; Sánchez-Sorzano, C.Ó.; et al. The Structural Role of SARS-CoV-2 Genetic Background in the Emergence and Success of Spike Mutations: The Case of the Spike A222V Mutation. PLoS Pathog. 2022, 18, e1010631. [Google Scholar] [CrossRef]
- Hu, L.; Tang, Y.; Mei, L.; Liang, M.; Huang, J.; Wang, X.; Wu, L.; Jiang, J.; Li, L.; Long, F.; et al. A New Intracellular Targeting Motif in the Cytoplasmic Tail of the Spike Protein May Act as a Target to Inhibit SARS-CoV-2 Assembly. Antivir. Res. 2023, 209, 105509. [Google Scholar] [CrossRef] [PubMed]
- Thomson, E.C.; Rosen, L.E.; Shepherd, J.G.; Spreafico, R.; da Silva Filipe, A.; Wojcechowskyj, J.A.; Davis, C.; Piccoli, L.; Pascall, D.J.; Dillen, J.; et al. Circulating SARS-CoV-2 Spike N439K Variants Maintain Fitness While Evading Antibody-Mediated Immunity. Cell 2021, 184, 1171–1187. [Google Scholar] [CrossRef]
- Wu, W.; Cheng, Y.; Zhou, H.; Sun, C.; Zhang, S. The SARS-CoV-2 Nucleocapsid Protein: Its Role in the Viral Life Cycle, Structure and Functions, and Use as a Potential Target in the Development of Vaccines and Diagnostics. Virol. J. 2023, 20, 6. [Google Scholar] [CrossRef] [PubMed]
- Moody, R.; Wilson, K.L.; Boer, J.C.; Holien, J.K.; Flanagan, K.L.; Jaworowski, A.; Plebanski, M. Predicted B Cell Epitopes Highlight the Potential for COVID-19 to Drive Self-Reactive Immunity. Front. Bioinform. 2021, 1, 709533. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, H. COVID-19: Attacks Immune Cells and Interferences With Antigen Presentation Through MHC-Like Decoy System. J. Immunother. 2023, 46, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Alsuwairi, F.A.; Alsaleh, A.N.; Alsanea, M.S.; Al-Qahtani, A.A.; Obeid, D.; Almaghrabi, R.S.; Alahideb, B.M.; AlAbdulkareem, M.A.; Mutabagani, M.S.; Althawadi, S.I.; et al. Association of SARS-CoV-2 Nucleocapsid Protein Mutations with Patient Demographic and Clinical Characteristics during the Delta and Omicron Waves. Microorganisms 2023, 11, 1288. [Google Scholar] [CrossRef]
- Wu, H.; Xing, N.; Meng, K.; Fu, B.; Xue, W.; Dong, P.; Tang, W.; Xiao, Y.; Liu, G.; Luo, H.; et al. Nucleocapsid Mutations R203K/G204R Increase the Infectivity, Fitness, and Virulence of SARS-CoV-2. Cell Host Microbe 2021, 29, 1788–1801. [Google Scholar] [CrossRef]
- Haddad, D.; John, S.E.; Mohammad, A.; Hammad, M.M.; Hebbar, P.; Channanath, A.; Nizam, R.; Al-Qabandi, S.; Madhoun, A.A.; Alshukry, A.; et al. SARS-CoV-2: Possible Recombination and Emergence of Potentially More Virulent Strains. PLoS ONE 2021, 16, e0251368. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, A.; Chaudhari, M.; Mahera, S.; Saiyed, Z.; Nathani, N.M.; Shukla, S.; Patel, D.; Patel, C.; Joshi, M.; Joshi, C.G. In-Silico Analysis Reveals Lower Transcription Efficiency of C241T Variant of SARS-CoV-2 with Host Replication Factors MADP1 and hnRNP-1. Inform. Med. Unlocked 2021, 25, 100670. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, R.S.M.A.; McLellan, A.D. Implications of SARS-CoV-2 Mutations for Genomic RNA Structure and Host microRNA Targeting. Int. J. Mol. Sci. 2020, 21, 4807. [Google Scholar] [CrossRef] [PubMed]
- Periwal, N.; Rathod, S.B.; Sarma, S.; Johar, G.S.; Jain, A.; Barnwal, R.P.; Srivastava, K.R.; Kaur, B.; Arora, P.; Sood, V. Time Series Analysis of SARS-CoV-2 Genomes and Correlations among Highly Prevalent Mutations. Microbiol. Spectr. 2022, 10, e01219-22. [Google Scholar] [CrossRef]
- Lopez, B.J.; Andrews, N.; Gower, C.; Gallagher, E.; Simmons, R.; Thelwall, S.; Stowe, J.; Tessier, E.; Groves, N.; Dabrera, G.; et al. Effectiveness of Covid-19 Vaccines against the B.1.617.2 (Delta) Variant. N. Engl. J. Med. 2021, 385, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Fowlkes, A.; Gaglani, M.; Groover, K.; Thiese, M.S.; Tyner, H.; Ellingson, K. Effectiveness of COVID-19 Vaccines in Preventing SARS-CoV-2 Infection Among Frontline Workers Before and During B.1.617.2 (Delta) Variant Predominance—Eight U.S. Locations, December 2020–August 2021. Morb. Mortal. Wkly. Rep. 2021, 70, 1167–1169. [Google Scholar] [CrossRef] [PubMed]
- Mengist, H.M.; Kombe, K.A.J.; Mekonnen, D.; Abebaw, A.; Getachew, M.; Jin, T. Mutations of SARS-CoV-2 Spike Protein: Implications on Immune Evasion and Vaccine-Induced Immunity. Semin. Immunol. 2021, 55, 101533. [Google Scholar] [CrossRef]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, Infectivity, and Neutralization of a Spike L452R SARS-CoV-2 Variant. Cell 2021, 184, 3426–3437. [Google Scholar] [CrossRef]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 Spike L452R Variant Evades Cellular Immunity and Increases Infectivity. Cell Host Microbe 2021, 29, 1124–1136. [Google Scholar] [CrossRef]
- Wall, E.C.; Wu, M.; Harvey, R.; Kelly, G.; Warchal, S.; Sawyer, C.; Daniels, R.; Hobson, P.; Hatipoglu, E.; Ngai, Y.; et al. Neutralising Antibody Activity against SARS-CoV-2 VOCs B.1.617.2 and B.1.351 by BNT162b2 Vaccination. Lancet 2021, 397, 2331–2333. [Google Scholar] [CrossRef]
- Niyonkuru, M.; Pedersen, R.M.; Assing, K.; Andersen, T.E.; Skov, M.N.; Johansen, I.S.; Madsen, L.W. Prolonged Viral Shedding of SARS-CoV-2 in Two Immunocompromised Patients, a Case Report. BMC Infect. Dis. 2021, 21, 743. [Google Scholar] [CrossRef]
- Hossain, M.E.; Lister, D.; Bartolo, C.; Kinsella, P.M.; Knox, J.; Aldrich, R.; Cowan, R.; Commons, R.J. Prolonged Viral Shedding in Patients with Mild to Moderate COVID-19 Disease: A Regional Perspective. Infect. Dis. 2021, 14, 11786337211010428. [Google Scholar] [CrossRef]
- Long, H.; Zhao, J.; Zeng, H.-L.; Lu, Q.-B.; Fang, L.-Q.; Wang, Q.; Wu, Q.-M.; Liu, W. Prolonged Viral Shedding of SARS-CoV-2 and Related Factors in Symptomatic COVID-19 Patients: A Prospective Study. BMC Infect. Dis. 2021, 21, 1282. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Triche, T.J.; Bard, J.D.; Biegel, J.A.; Judkins, A.R.; Gai, X. Spike Protein NTD Mutation G142D in SARS-CoV-2 Delta VOC Lineages Is Associated with Frequent Back Mutations, Increased Viral Loads, and Immune Evasion. Medrxiv 2021. preprint. [Google Scholar] [CrossRef]
- Siedner, M.J.; Boucau, J.; Gilbert, R.F.; Uddin, R.; Luu, J.; Haneuse, S.; Vyas, T.; Reynolds, Z.; Iyer, S.; Chamberlin, G.C.; et al. Duration of Viral Shedding and Culture Positivity with Postvaccination SARS-CoV-2 Delta Variant Infections. JCI Insight 2022, 7, e155483. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Yun, K.W.; Jeong, H.; Kim, B.; Kim, M.J.; Park, J.H.; Shin, H.S.; Oh, H.S.; Sung, H.; Song, M.G.; et al. SARS-CoV-2 Shedding Dynamics and Transmission in Immunosuppressed Patients. Virulence 2022, 13, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Ling-Hu, T.; Rios-Guzman, E.; Lorenzo-Redondo, R.; Ozer, E.A.; Hultquist, J.F. Challenges and Opportunities for Global Genomic Surveillance Strategies in the COVID-19 Era. Viruses 2022, 14, 2532. [Google Scholar] [CrossRef] [PubMed]
- Silk, B.J. COVID-19 Surveillance After Expiration of the Public Health Emergency Declaration—United States, May 11, 2023. MMWR Morb. Mortal. Wkly. Rep. 2023, 72, 523. [Google Scholar] [CrossRef]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.-Y. Impact of BA.1, BA.2, and BA.4/BA.5 Omicron Mutations on Therapeutic Monoclonal Antibodies. Comput. Biol. Med. 2023, 167, 107576. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Observation (n = 292) |
|---|---|
| Age [years, median (IQR)] | 32.0 (26.0–45.0) |
| Age group [n (%)] | |
| Children and teens (<18 years) | 14 (4.8%) |
| Young adults (18–40 years) | 179 (61.3%) |
| Adults (41–65 years) | 85 (29.1%) |
| Elderly (>65 years) | 14 (4.8%) |
| Sex [n (%)] | |
| Female | 140 (47.9%) |
| Male | 152 (52.1%) |
| Hospital location [n (%)] | |
| Bangka Belitung | 35 (12.0%) |
| Central Kalimantan | 52 (17.8%) |
| East Nusa Tenggara | 23 (7.9%) |
| Greater Jakarta (Jabodetabek) | 58 (19.9%) |
| Jambi | 38 (13.0%) |
| Maluku | 36 (12.3%) |
| North Sulawesi | 32 (11.0%) |
| West Java | 18 (6.16%) |
| Sample type [n (%)] | |
| Nasopharyngeal swab | 292 (100%) |
| Collection month [n (%)] | |
| January | 43 (14.7%) |
| February | 45 (15.4%) |
| March | 9 (3.1%) |
| April | 1 (0.3%) |
| May | 4 (1.4%) |
| June | 53 (18.2%) |
| July | 120 (41.1%) |
| August | 16 (5.5%) |
| October | 1 (0.3%) |
| Hospitalized [n (%)] | |
| Yes | 56 (19.2%) |
| Unknown | 236 (80.8%) |
| Other conditions [n (%)] | |
| Breakthrough infection 1 | 32 (11.0%) |
| Prolonged viral shedding 2 | 5 (1.7%) |
| Reinfection | 1 (0.3%) |
| Breakthrough infection and prolonged viral shedding | 3 (1.0%) |
| Breakthrough infection and reinfection | 3 (1.0%) |
| PCR cycle threshold (Ct) value [median (IQR)] | |
| N gene | 18.3 (15.4–21.9) |
| ORF1ab gene | 16.7 (14.7–20.4) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oktavianthi, S.; Lages, A.C.; Kusuma, R.; Kurniasih, T.S.; Trimarsanto, H.; Andriani, F.; Rustandi, D.; Meriyanti, T.; Yusuf, I.; Malik, S.G.; et al. Whole-Genome Sequencing and Mutation Analyses of SARS-CoV-2 Isolates from Indonesia. Pathogens 2024, 13, 279. https://doi.org/10.3390/pathogens13040279
Oktavianthi S, Lages AC, Kusuma R, Kurniasih TS, Trimarsanto H, Andriani F, Rustandi D, Meriyanti T, Yusuf I, Malik SG, et al. Whole-Genome Sequencing and Mutation Analyses of SARS-CoV-2 Isolates from Indonesia. Pathogens. 2024; 13(4):279. https://doi.org/10.3390/pathogens13040279
Chicago/Turabian StyleOktavianthi, Sukma, Aksar Chair Lages, Rinaldy Kusuma, Tri Shinta Kurniasih, Hidayat Trimarsanto, Febi Andriani, David Rustandi, Tandry Meriyanti, Irawan Yusuf, Safarina G. Malik, and et al. 2024. "Whole-Genome Sequencing and Mutation Analyses of SARS-CoV-2 Isolates from Indonesia" Pathogens 13, no. 4: 279. https://doi.org/10.3390/pathogens13040279
APA StyleOktavianthi, S., Lages, A. C., Kusuma, R., Kurniasih, T. S., Trimarsanto, H., Andriani, F., Rustandi, D., Meriyanti, T., Yusuf, I., Malik, S. G., Jo, J., & Suriapranata, I. (2024). Whole-Genome Sequencing and Mutation Analyses of SARS-CoV-2 Isolates from Indonesia. Pathogens, 13(4), 279. https://doi.org/10.3390/pathogens13040279