
Lipid Profile and Cardiovascular Risk Modification after Hepatitis C Virus Eradication

 ,
,  and
and
Abstract
1. Introduction
2. Epidemiology of HCV Infection and Clinical Course
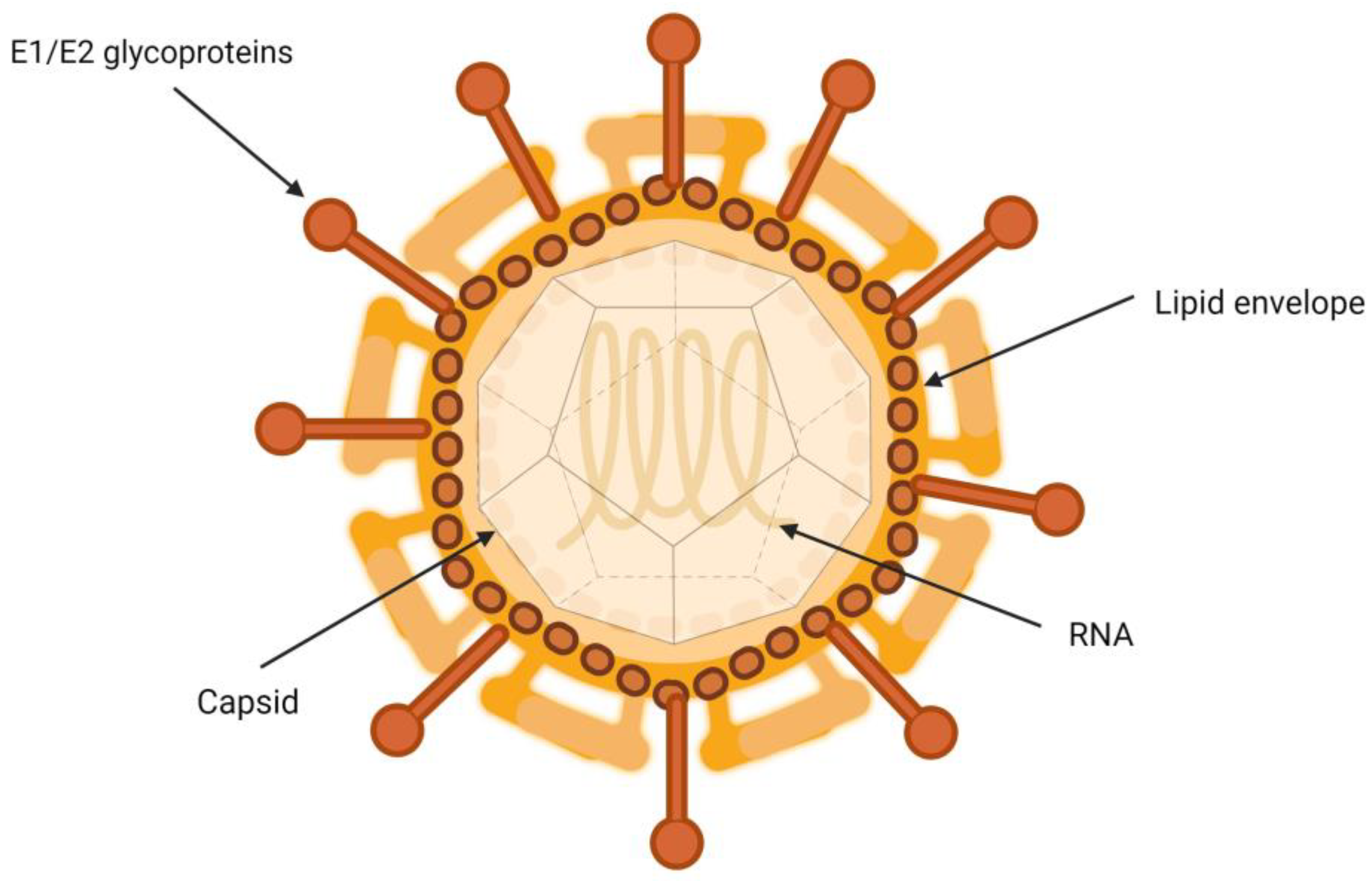
3. Biology of Hepatitis C Virus and Its Association with Lipoproteins
Characteristics of the Hepatitis C Virus
4. Lipoproteins
4.1. Exogenous Pathway of Lipoprotein Metabolism
4.2. Endogenous Pathway of Lipoprotein Metabolism
5. Lipoprotein Profile Assessment
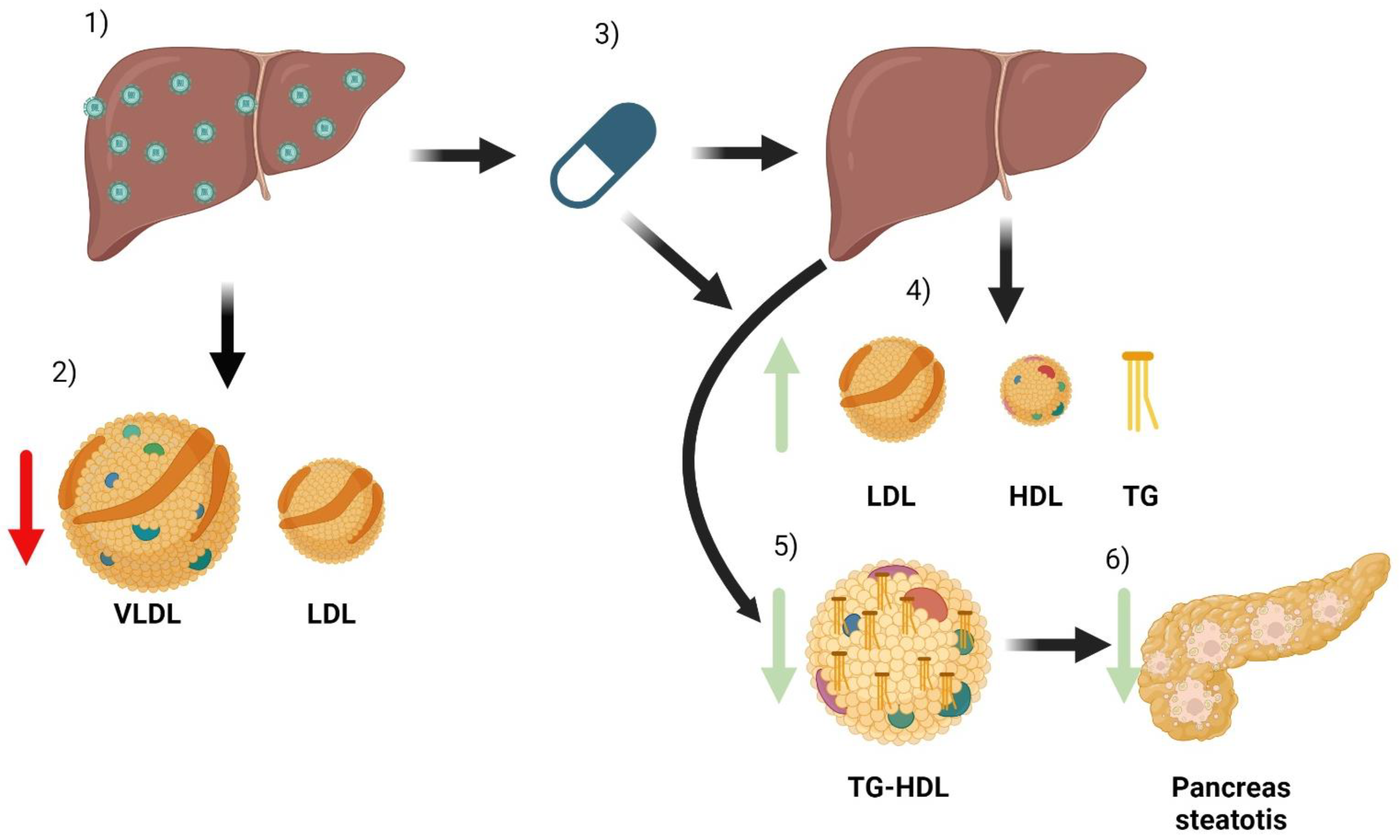
6. Alterations in Lipid Metabolism Associated with HCV Infection
6.1. Diabetes Mellitus and Insulin Resistance
6.2. Cardiovascular Diseases
7. Metabolic Changes Related to Treatment with Direct-Acting Antivirals

{kind=link}
{kind=link}
| Study | Year | N | Design | Follow-Up | Drug | Genotype | Cirrhosis | Total Cholesterol | HDL | LDL | TG | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chida T [79] | 2018 | 70 | Retrospective | 4 weeks | DCL + ASV | 1b | NA | ↑↑ | ↑ | ↑↑ | NA | |
| Endo [80] | 2014–2016 | 276 | Retrospective | 24 weeks | DCV + ASV SOF + LDV SOF/LDV | 1b | NA | ↑↑ | ↑ | ↑↑ | NA | |
| Inoue [67] | 2018 | 216 | Prospective post Hoc | 48 weeks | DCV + ASV SOF/RBV | 1b y 2 | NA | ↑↑ | ↑ | = | = | |
| Meissner [66] | 2015 | 54 | Retrospective | 24 weeks | SOF/RBV | 1 | NA | NA | NA | ↑↑ | ↓ | |
| Chaudhury [81] | 2011–2017 | 251 | Prospective Ad hoc | 28 months | NA | 1 | NA | NA | NA | ↑↑ | ↓ | 30% HIV |
| Sun [70] | 2018 | 24 | Prospective | ND | EBV/GPV LDV/SOF | 1 | NA | ↑ | NA | NA | ↓ | |
| Townsend [82] | 2016 | 90 | Prospective post Hoc | 24 weeks | LDV/SOF SOF/LDV | 1 | NA | NA | NA | ↑↑ | NA | 60% HIV |
| Morales [69] | 2014–2016 | 52 | Retrospective | 6 months | SOF IFN LDV SIM/RBV | All | 24% | ↑↑ | ↓ | ↑↑ | ↓ | |
| Beig [83] | 1998–2016 | 132 | Retrospective | 48 weeks | DAA without IFN | All | No | ↑↑ | NA | ↑↑ | NA | Transplanted |
| Carvalho [84] | 2018 | 178 | Prospective | ND | DAA without IFN or RBV | All | NA | ↑↑ | NA | ↑↑ | ↓ | |
| Gitto [76] | 2015 | 100 | Prospective | 24 weeks | DAA + RBV | All | 80% | ↑↑ | NA | NA | NA | |
| Mauss [68] | 2017 | 520 | Prospective | ND | DAA | All | NA | ↑↑ | NA | ↑↑ | = | |
| El Sagheer [85] | 2018 | 80 | Retrospective | ND | SIM/SOF | 4 | >50% | ↑↑ | ↑ | ↑↑ | ↓ | |
| Gonzalez Colominas [86] | 2019 | 226 | Prospective | 48 weeks | DAA | All | 50% | ↑↑ | ↑ | ↑↑ | NA | |
| Doyle [87] | 2015–2016 | 24 | Prospective | 24 weeks | OBV/DSV | All | NA | ↑↑ | ↑ | NA | ↑ | Evaluation of APOA, APOB and APOE, HOMA-IR |
| Ichikawa [88] | 2014–2016 | 39 | Prospective | 24 weeks | DCV/ASV | 1b | NA | ↑↑ | = | ↑↑ | = | |
| Shimizu [89] | 2012–2016 | 70 | Retrospective | 48 weeks | All | 1 y 2 | NA | = | ↑ | ↑↑ | = | All patients with steatosis |
| Cheng [90] | 2017 | 102 | Prospective | 12 weeks | All | All (1b 80%) | 75% | ↑↑ | = | ↑↑ | ↑ | |
| Jain [91] | 2017 | 50 | Prospective | 12 weeks | SOF/DCV | NC | NA | ↑↑ | = | ↑↑ | = | |
| Petta [92] | 2018 | 182 | Prospective | 48 weeks | All | All | 66% | ↑↑ | NA | NA | NA | |
| Casas-Deza [78] | 2019 | 177 | Prospective | 48 weeks | All | All | 10% | ↑↑ | ↑ | ↑↑ | ↑ |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Simmonds, P. The origin of hepatitis C virus. Curr. Top. Microbiol. Immunol. 2013, 369, 1–15. [Google Scholar]
- Suryaprasad, A.G.; White, J.Z.; Xu, F.; Eichler, B.A.; Hamilton, J.; Patel, A.; Bel-Hamdounia, S.; R-Church, D.; Barton, K.; Fisher, C.; et al. Emerging epidemic of hepatitis C virus infections among young nonurban persons who inject drugs in the United States, 2006–2012. Clin. Infect. Dis. 2014, 59, 1411–1419. [Google Scholar] [CrossRef]
- Zein, N.N. Clinical significance of hepatitis C virus genotypes. Clin. Microbiol. Rev. 2000, 13, 223–235. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Hepatitis Report 2017; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Polaris Observatory HCV Collaborators. Global change in hepatitis C virus prevalence and cascade of care between 2015 and 2020: A modelling study. The lancet. Gastroenterol. Hepatol. 2022, 7, 396–415. [Google Scholar] [CrossRef]
- Frieden, T.R.; Harold Jaffe, D.W.; Rasmussen, S.A.; Leahy, M.A.; Martinroe, J.C.; Spriggs, S.R.; Doan, Q.M.; King Terraye MStarr, P.H.; Roper, W.L.; Hill, C.; et al. Sexually Transmitted Diseases Treatment Guidelines. Morb. Mortal Wkly Rep. 2015, 64, 1–140. [Google Scholar]
- Catanese, M.T.; Uryu, K.; Kopp, M.; Edwards, T.J.; Andrus, L.; Rice, W.J.; Silvestry, M.; Kuhn, R.J.; Rice, C.M. Ultrastructural analysis of hepatitis C virus particles. Proc. Natl. Acad. Sci. USA 2013, 110, 9505–9510. [Google Scholar] [CrossRef]
- Vieyres, G.; Dubuisson, J.; Pietschmann, T. Incorporation of Hepatitis C Virus E1 and E2 Glycoproteins: The Keystones on a Peculiar Virion. Viruses 2014, 6, 1149–1187. [Google Scholar] [CrossRef]
- André, P.; Komurian-Pradel, F.; Deforges, S.; Perret, M.; Berland, J.L.; Sodoyer, M.; Pol, S.; Chébrot, C.; Paranhos-Baccalà, G.; Lotteau, V. Characterization of Low- and Very-Low-Density Hepatitis C Virus RNA-Containing Particles. J. Virol. 2002, 76, 6919–6928. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Penin, F.; Lohmann, V.; André, P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 2011, 19, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D. Virion assembly and release. Curr. Top. Microbiol. Immunol. 2013, 369, 199–218. [Google Scholar] [CrossRef]
- Pileri, P.; Uematsu, Y.; Campagnoli, S.; Galli, G.; Falugi, F.; Petracca, R.; Weiner, A.J.; Houghton, M.; Rosa, D.; Grandi, G.; et al. Binding of hepatitis C virus to CD81. Science 1998, 282, 938–941. [Google Scholar] [CrossRef]
- Lohmann, V. Hepatitis C virus RNA replication. Curr. Top. Microbiol. Immunol. 2013, 369, 167–198. [Google Scholar] [CrossRef]
- Ferraris, P.; Blanchard, E.; Roingeard, P. Ultrastructural and biochemical analyses of hepatitis C virus-associated host cell membranes. J. Gen. Virol. 2010, 91, 2230–2237. [Google Scholar] [CrossRef]
- Paul, D.; Hoppe, S.; Saher, G.; Krijnse-Locker, J.; Bartenschlager, R. Morphological and Biochemical Characterization of the Membranous Hepatitis C Virus Replication Compartment. 2013. Available online: http://jvi.asm.org/ (accessed on 10 January 2024).
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010, 6, e1000719. [Google Scholar] [CrossRef]
- Targett-Adams, P.; Boulant, S.; McLauchlan, J. Visualization of Double-Stranded RNA in Cells Supporting Hepatitis C Virus RNA Replication. J. Virol. 2008, 82, 2182–2195. [Google Scholar] [CrossRef]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef]
- Peet, D.J.; Turley, S.D.; Ma, W.; Janowski, B.A.; Lobaccaro, J.M.; Hammer, R.E.; Mangelsdorf, D.J. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell 1998, 93, 693–704. [Google Scholar] [CrossRef]
- O’Connell, B.J.; Denis, M.; Genest, J. Cellular physiology of cholesterol efflux in vascular endothelial cells. Circulation 2004, 110, 2881–2888. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R. Lipid and Lipoprotein Metabolism. Endocrinol. Metab. Clin. N. Am. 2022, 51, 437–458. [Google Scholar] [CrossRef] [PubMed]
- Paunovska, K.; Da Silva Sanchez, A.J.; Lokugamage, M.P.; Loughrey, D.; Echeverri, E.S.; Cristian, A.; Hatit, M.Z.C.; Santangelo, P.J.; Zhao, K.; Dahlman, J.E. The Extent to Which Lipid Nanoparticles Require Apolipoprotein E and Low-Density Lipoprotein Receptor for Delivery Changes with Ionizable Lipid Structure. Nano Lett. 2022, 22, 10025–10033. [Google Scholar] [CrossRef] [PubMed]
- Gugliucci, A. Triglyceride-Rich Lipoprotein Metabolism: Key Regulators of Their Flux. J. Clin. Med. 2023, 12, 4399. [Google Scholar] [CrossRef]
- Wang, N.; Silver, D.L.; Costet, P.; Tall, A.R. Specific Binding of ApoA-I, Enhanced Cholesterol Efflux, and Altered Plasma Membrane Morphology in Cells Expressing ABC1*. 2000. Available online: http://www.jbc.org (accessed on 11 January 2024).
- Giammanco, A.; Cefalù, A.B.; Noto, D.; Averna, M.R. The pathophysiology of intestinal lipoprotein production. Front. Physiol. 2015, 6, 61. [Google Scholar] [CrossRef]
- Ferrari, R.; Aguiar, C.; Alegria, E.; Bonadonna, R.C.; Cosentino, F.; Elisaf, M.; Farnier, M.; Ferrières, J.; Filardi, P.P.; Hancu, N.; et al. Current practice in identifying and treating cardiovascular risk, with a focus on residual risk associated with atherogenic dyslipidaemia. Eur. Heart J. Suppl. J. Eur. Soc. Cardiol. 2016, 18 (Suppl. SC), C2–C12. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Brewer, H.B., Jr.; Fazio, S.; Hussain, M.M.; Kontush, A.; Krauss, R.M.; Otvos, J.D.; Remaley, A.T.; Schaefer, E.J. HDL measures, particle heterogeneity, proposed nomenclature, and relation to atherosclerotic cardiovascular events. Clin. Chem. 2011, 57, 392–410. [Google Scholar] [CrossRef] [PubMed]
- Fruchart, J.C.; Davignon, J.; Hermans, M.P.; Al-Rubeaan, K.; Amarenco, P.; Assmann, G.; Barter, P.; Betteridge, J.; Bruckert, E.; Cuevas, A.; et al. Residual macrovascular risk in 2013: What have we learned? Cardiovasc. Diabetol. 2014, 13, 28. [Google Scholar] [CrossRef] [PubMed]
- Fruchart, J.C.; Sacks, F.M.; Hermans, M.P.; Assmann, G.; Brown, W.V.; Ceska, R.; Chapman, M.J.; Dodson, P.M.; Fioretto, P.; Ginsberg, H.N.; et al. The Residual Risk Reduction Initiative: A call to action to reduce residual vascular risk in dyslipidaemic patient. Diabetes Vasc. Dis. Res. 2008, 5, 319–335. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Underberg, J.A. Systematic review: Evaluating the effect of lipid-lowering therapy on lipoprotein and lipid values. Cardiovasc. Drugs Ther. 2013, 27, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Williams, K.J.; Borén, J. Subendothelial Lipoprotein Retention as the Initiating Process in Atherosclerosis. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef]
- Borén, J.; Williams, K.J. The central role of arterial retention of cholesterolrich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: A triumph of simplicity. Curr Opin Lipidol. 2016, 27, 473–483. [Google Scholar] [CrossRef]
- Pintó, X.; Masana, L.; Civeira, F.; Real, J.; Ibarretxe, D.; Candas, B.; Puzo, J.; Díaz, J.; Amigó, N.; Esteban, M.; et al. Documento de consenso de un grupo de expertos de la Sociedad Española de Arteriosclerosis (SEA) sobre el uso clínico de la resonancia magnética nuclear en el estudio del metabolismo lipoproteico (Liposcale). Clínica Investig. Arterioscler. 2020, 32, 219–229. [Google Scholar] [CrossRef]
- Otvos, J.D.; Mora, S.; Shalaurova, I.; Greenland, P.; MacKey, R.H.; Goff, D.C. Clinical implications of discordance between low-density lipoprotein cholesterol and particle number. J. Clin. Lipidol. 2011, 5, 105–113. [Google Scholar] [CrossRef]
- Mora, S.; Caulfield, M.P.; Wohlgemuth, J.; Chen, Z.; Superko, H.R.; Rowland, C.M.; Glynn, R.J.; Ridker, P.M.; Krauss, R.M. Atherogenic Lipoprotein Subfractions Determined by Ion Mobility and First Cardiovascular Events After Random Allocation to High-Intensity Statin or Placebo: The Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) Trial. Circulation 2015, 132, 2220–2229. [Google Scholar] [CrossRef][Green Version]
- Bermudez-Lopez, M.; Perpiñan, H.; Amigo, N.; Castro, E.; Alonso, N.; Mauricio, D.; Fernandez, E.; Valdivieso, J.M. Advanced Lipoprotein Parameters Could Better Explain Atheromatosis in Non-Diabetic Chronic Kidney Disease Patients. Available online: https://academic.oup.com/ckj/article/14/12/2591/6316255 (accessed on 10 January 2023).
- Tehrani, D.M.; Zhao, Y.; Blaha, M.J.; Mora, S.; Mackey, R.H.; Michos, E.D.; Budoff, M.J.; Cromwell, W.; Otvos, J.D.; Rosenblit, P.D.; et al. Discordance of Low-Density Lipoprotein and High-Density Lipoprotein Cholesterol Particle Versus Cholesterol Concentration for the Prediction of Cardiovascular Disease in Patients with Metabolic Syndrome and Diabetes Mellitus (from the Multi-Ethnic Study of Atherosclerosis [MESA]). Am. J. Cardiol. 2016, 117, 1921–1927. [Google Scholar] [CrossRef]
- Cantey, E.P.; Wilkins, J.T. Discordance between lipoprotein particle number and cholesterol content: An update. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 25, 130–136. [Google Scholar] [CrossRef]
- Gumber, S.C.; Chopra, S. Hepatitis C: A multifaceted disease-Review of extrahepatic manifestations. Ann. Intern. Med. 1995, 123, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Zignego, A.L.; Giannini, C.; Monti, M.; Gragnani, L. Hepatitis C virus lymphotropism: Lessons from a decade of studies. Dig. Liver Dis. 2007, 39 (Suppl. S1), S38–S45. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Liu, R.; Pan, Q.; Wang, G.; Cheng, D.; Yang, J.; Chen, H.; Xu, G. De novo lipogenesis is elicited dramatically in human hepatocellular carcinoma especially in hepatitis C virus-induced hepatocellular carcinoma. MedComm 2020, 1, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Kang, R.; Zhao, Z. Is Hepatitis C Associated with Atherosclerotic Burden? A Systematic Review and Meta-Analysis. Available online: www.plosone.org (accessed on 29 December 2023).
- Bassendine, M.F.; Nielsen, S.U.; Bridge, S.H.; Felmlee, D.J.; Sheridan, D.A.; Packard, C.J.; Neely, R.D. Hepatitis C virus and atherosclerosis: A legacy after virologic cure? Clin. Res. Hepatol. Gastroenterol. 2017, 41, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, T.; Miyaaki, H.; Miuma, S.; Motoyoshi, Y.; Narita, S.; Toda, S.; Takahashi, Y.; Honda, T.; Yajima, H.; Uehara, R.; et al. Carotid Intima-media Thickness and Small Dense Low-density Lipoprotein Cholesterol Increase after One Year of Treatment with Direct-acting Antivirals in Patients with Hepatitis C Virus Infection. Intern Med. 2019, 58, 1209–1215. [Google Scholar] [CrossRef]
- Drazilova, S.; Gazda, J.; Janicko, M.; Jarcuska, P. Chronic Hepatitis C Association with Diabetes Mellitus and Cardiovascular Risk in the Era of DAA Therapy. Can. J. Gastroenterol. Hepatol. 2018, 2018, 6150861. [Google Scholar] [CrossRef]
- Huang, J.F.; Huang, C.F.; Yeh, M.L.; Dai, C.Y.; Yu, M.L.; Chuang, W.L. Updates in the management and treatment of HCV genotype 3, what are the remaining challenges? Expert Rev. Anti-Infect. Ther. 2018, 16, 907–912. [Google Scholar] [CrossRef]
- Chan, A.; Patel, K.; Naggie, S. Genotype 3 Infection: The Last Stand of Hepatitis C Virus. Drugs 2017, 77, 131–144. [Google Scholar] [CrossRef]
- Serfaty, L.; Capeau, J. Hepatitis C, insulin resistance and diabetes: Clinical and pathogenic data. Liver Int. 2009, 29 (Suppl. S2), 13–25. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.Y.; Yeh, M.L.; Huang, C.F.; Hou, C.H.; Hsieh, M.Y.; Huang, J.F.; Lin, I.L.; Lin, Z.Y.; Chen, S.C.; Wang, L.Y.; et al. Chronic hepatitis C infection is associated with insulin resistance and lipid profiles. J. Gastroenterol. Hepatol. 2015, 30, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Desbois, A.C.; Cacoub, P. Diabetes mellitus, insulin resistance and hepatitis C virus infection: A contemporary review. World J. Gastroenterol. 2017, 23, 1697. [Google Scholar] [CrossRef] [PubMed]
- Drazilova, S.; Janicko, M.; Skladany, L.; Kristian, P.; Oltman, M.; Szantova, M.; Krkoska, D.; Mazuchova, E.; Piesecka, L.; Vahalova, V.; et al. Glucose Metabolism Changes in Patients with Chronic Hepatitis C Treated with Direct Acting Antivirals. Can. J. Gastroenterol. Hepatol. 2018, 2018, 6095097. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Y.; Fujie, H.; Miyoshi, H.; Tsutsumi, T.; Tsukamoto, K.; Kimura, S.; Moriya, K.; Koike, K. Hepatitis C Virus Infection and Diabetes: Direct Involvement of the Virus in the Development of Insulin Resistance. Gastroenterology 2004, 126, 840–848. [Google Scholar] [CrossRef]
- Thompson, A.J.; Patel, K.; Chuang, W.L.; Lawitz, E.J.; Rodriguez-Torres, M.; Rustgi, V.K.; Flisiak, R.; Pianko, S.; Diago, M.; Arora, S.; et al. Viral clearance is associated with improved insulin resistance in genotype 1 chronic hepatitis C but not genotype 2/3. Gut 2012, 61, 128. [Google Scholar] [CrossRef]
- Romero-Gómez, M.; Del Mar Viloria, M.; Andrade, R.J.; Salmerón, J.; Diago, M.; Fernández-Rodríguez, C.M.; Corpas, R.; Cruz, M.; Grande, L.; Vázquez, L.; et al. Insulin resistance impairs sustained response rate to peginterferon plus ribavirin in chronic hepatitis C patients. Gastroenterology 2005, 128, 636–641. [Google Scholar] [CrossRef]
- Li, J.; Gordon, S.C.; Rupp, L.B.; Zhang, T.; Trudeau, S.; Holmberg, S.D.; Moorman, A.C.; Spradling, P.R.; Teshale, E.H.; Boscarino, J.A.; et al. Sustained virological response does not improve long-term glycaemic control in patients with type 2 diabetes and chronic hepatitis C. Liver Int. Off. J. Int. Assoc. Study Liver 2019, 39, 1027–1032. [Google Scholar] [CrossRef]
- Nevola, R.; Rinaldi, L.; Zeni, L.; Sasso, F.C.; Pafundi, P.C.; Guerrera, B.; Marrone, A.; Giordano, M.; Adinolfi, L.E. Metabolic and renal changes in patients with chronic hepatitis C infection after hepatitis C virus clearance by direct-acting antivirals. JGH Open Open Access J. Gastroenterol. Hepatol. 2020, 4, 713–721. [Google Scholar] [CrossRef]
- Arase, Y.; Suzuki, F.; Suzuki, Y.; Akuta, N.; Kobayashi, M.; Kawamura, Y.; Yatsuji, H.; Sezaki, H.; Hosaka, T.; Hirakawa, M.; et al. Sustained Virological Response Reduces Incidence of Onset of Type 2 Diabetes in Chronic Hepatitis C. 2008. Available online: https://aasldpubs.onlinelibrary.wiley.com/doi/10.1002/hep.22703 (accessed on 20 January 2024).
- Voulgaris, T.; Sevastianos, V.A. Atherosclerosis as Extrahepatic Manifestation of Chronic Infection with Hepatitis C Virus. Hepat. Res. Treat. 2016, 2016, 7629318. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Boddi, M.; Abbate, R.; Chellini, B.; Giusti, B.; Solazzo, V.; Soft, F.; Pratesi, G.; Pratesi, C.; Gensini, G.; Zignego, A.L. HCV infection facilitates asymptomatic carotid atherosclerosis: Preliminary report of HCV RNA localization in human carotid plaques. Dig. Liver Dis. 2007, 39 (Suppl. S1), S55–S60. [Google Scholar] [CrossRef]
- Adinolfi, L.E.; Zampino, R.; Restivo, L.; Lonardo, A.; Guerrera, B.; Marrone, A.; Nascimbeni, F.; Florio, A.; Loria, P. Chronic hepatitis C virus infection and atherosclerosis: Clinical impact and mechanisms. World J. Gastroenterol. WJG 2014, 20, 3410. [Google Scholar] [CrossRef] [PubMed]
- Wu, V.C.C.; Chen, T.H.; Wu, M.; Cheng, C.W.; Chen, S.W.; Chang, C.W.; Chen, C.C.; Chang, S.H.; Hung, K.C.; Chern, M.S.; et al. Comparison of cardiovascular outcomes and all-cause mortality in patients with chronic hepatitis B and C: A 13-year nationwide population-based study in Asia. Atherosclerosis 2018, 269, 178–184. [Google Scholar] [CrossRef]
- Maruyama, S.; Koda, M.; Oyake, N.; Sato, H.; Fujii, Y.; Horie, Y.; Murawaki, Y. Myocardial injury in patients with chronic hepatitis C infection. J. Hepatol. 2013, 58, 11–15. [Google Scholar] [CrossRef]
- Rodríguez-Osorio, I.; Cid, P.; Morano, L.; Castro, Á.; Suárez, M.; Delgado, M.; Margusino, L.; Meijide, H.; Pernas, B.; Tabernilla, A.; et al. Real life experience with direct-acting antivirals agents against hepatitis C infection in elderly patients. J. Clin. Virol. 2017, 88, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Ottman, A.A.; Townsend, M.L.; Hashem, M.G.; DiMondi, V.P.; Britt, R.B. Incidence of Drug Interactions Identified by Clinical Pharmacists in Veterans Initiating Treatment for Chronic Hepatitis C Infection. Ann. Pharmacother. 2018, 52, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.H.; Tseng, C.W.; Lee, C.H.; Tseng, K.C. Drug-drug interactions between direct-acting antivirals and statins in the treatment of chronic hepatitis C. Tzu Chi Med. J. 2020, 32, 331–338. [Google Scholar]
- Meissner, E.G.; Lee, Y.J.; Osinusi, A.; Sims, Z.; Qin, J.; Sturdevant, D.; McHutchison, J.; Subramanian, M.; Sampson, M.; Naggie, S.; et al. Effect of Sofosbuvir and Ribavirin Treatment on Peripheral and Hepatic Lipid Metabolism in Chronic HCV, Genotype-1 Infected Patients. Hepatology 2015, 61, 790. [Google Scholar] [CrossRef]
- Inoue, T.; Goto, T.; Iio, E.; Matsunami, K.; Fujiwara, K.; Shinkai, N.; Matsuura, K.; Matsui, T.; Nojiri, S.; Tanaka, Y. Changes in serum lipid profiles caused by three regimens of interferon-free direct-acting antivirals for patients infected with hepatitis C virus. Hepatol. Res. 2018, 48, E203–E212. [Google Scholar] [CrossRef] [PubMed]
- Mauss, S.; Berger, F.; Wehmeyer, M.H.; Ingiliz, P.; Hueppe, D.; Lutz, T.; Simon, K.G.; Schewe, K.; Rockstroh, J.K.; Baumgarten, A.; et al. Effect of Antiviral Therapy for HCV on Lipid Levels. Antivir. Ther. 2017, 22, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Morales, A.L.; Junga, Z.; Singla, M.B.; Sjogren, M.; Torres, D.; Singal, M.B.; Sjogren, M.; Torres, D. Hepatitis C eradication with Sofosbuvir Leads to Significant Metabolic Changes. Retrosp. Cohort Study 2016, 8, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.Y.; Cheng, P.N.; Tseng, C.Y.; Tsai, W.J.; Chiu, Y.C.; Young, K.C. Favouring modulation of circulating lipoproteins and lipid loading capacity by direct antiviral agents grazoprevir/elbasvir or ledipasvir/sofosbuvir treatment against chronic HCV infection. Gut 2018, 67, 1342–1350. [Google Scholar] [CrossRef] [PubMed]
- Girona, J.; Amigó, N.; Ibarretxe, D.; Plana, N.; Rodríguez-Borjabad, C.; Heras, M.; Ferré, R.; Gil, M.; Correig, X.; Masana, L. HDL Triglycerides: A New Marker of Metabolic and Cardiovascular Risk. Available online: www.mdpi.com/journal/ijms (accessed on 25 January 2024).
- Bartolomei, G.; Cevik, R.E.; Marcello, A. Modulation of hepatitis C virus replication by iron and hepcidin in Huh7 hepatocytes. J. Gen. Virol. 2011, 92, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Petit, J.M.; Bour, J.B.; Galland-Jos, C.; Minello, A.; Verges, B.; Guiguet, M.; Brun, J.M.; Hillon, P. Risk factors for diabetes mellitus and early insulin resistance in chronic hepatitis C. J. Hepatol. 2001, 35, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.Y.; Chuang, W.L.; Ho, C.K.; Hsieh, M.Y.; Huang, J.F.; Lee, L.P.; Hou, N.J.; Lin, Z.Y.; Chen, S.C.; Hsieh, M.Y.; et al. Associations between hepatitis C viremia and low serum triglyceride and cholesterol levels: A community-based study. J. Hepatol. 2008, 49, 9–16. [Google Scholar] [CrossRef]
- Kim, A.Y. Clinical Infectious Diseases AASLD-IDSA HCV Guidance Panel a. Available online: www.HCVGuidelines.org (accessed on 12 December 2023).
- Gitto, S.; Cicero, A.F.G.; Loggi, E.; Giovannini, M.; Conti, F.; Grandini, E.; Guarneri, V.; Scuteri, A.; Vitale, G.; Cursaro, C.; et al. Worsening of Serum Lipid Profile after Direct Acting Antiviral Treatment. Ann. Hepatol. 2018, 17, 64–75. [Google Scholar] [CrossRef]
- Huang, C.F.; Dai, C.Y.; Yeh, M.L.; Huang, C.I.; Lee, H.C.; Lai, W.T.; Liang, P.C.; Lin, Y.H.; Hsieh, M.Y.; Hou, N.J.; et al. Cure or curd: Modification of lipid profiles and cardio-cerebrovascular events after hepatitis C virus eradication. Kaohsiung J. Med. Sci. 2020, 36, 920–928. [Google Scholar] [CrossRef]
- Casas-Deza, D.; Martínez-Sapiña, A.; Espina, S.; Garcia-Rodriguez, B.; Fernandez-Bonilla, E.M.; Sanz-Paris, A.; Gonzalez-Irazabal, Y.; Bernal-Monterde, V.; Arbones-Mainar, J.M. Evaluation of Cardiovascular Risk Factors after Hepatitis C Virus Eradication with Direct-Acting Antivirals in a Cohort of Treatment-Naïve Patients without History of Cardiovascular Disease. J. Clin. Med. 2022, 11, 4049. [Google Scholar] [CrossRef]
- Chida, T.; Kawata, K.; Ohta, K.; Matsunaga, E.; Ito, J.; Shimoyama, S.; Yamazaki, S.; Noritake, H.; Suzuki, T.; Suda, T.; et al. Rapid Changes in Serum Lipid Profiles during Combination Therapy with Daclatasvir and Asunaprevir in Patients Infected with Hepatitis C Virus Genotype 1b. Gut Liver 2018, 12, 201–207. [Google Scholar] [CrossRef]
- Endo, D.; Satoh, K.; Shimada, N.; Hokari, A.; Aizawa, Y. Impact of interferon-free antivirus therapy on lipid profiles in patients with chronic hepatitis C genotype 1b. World J. Gastroenterol. 2017, 23, 2355–2364. [Google Scholar] [CrossRef][Green Version]
- Chaudhury, C.S.; Sheehan, J.; Chairez, C.; Akoth, E.; Gross, C.; Silk, R.; Kattakuzhy, S.; Rosenthal, E.; Kottilil, S.; Masur, H.; et al. No Improvement in Hemoglobin A1c Following Hepatitis C Viral Clearance in Patients with and without HIV. J. Infect. Dis. 2017, 217, 47–50. [Google Scholar] [CrossRef]
- Townsend, K.; Meissner, E.G.; Sidharthan, S.; Sampson, M.; Remaley, A.T.; Tang, L.; Kohli, A.; Osinusi, A.; Masur, H.; Kottilil, S. Interferon-Free Treatment of Hepatitis C Virus in HIV/Hepatitis C Virus-Coinfected Subjects Results in Increased Serum Low-Density Lipoprotein Concentration. AIDS Res. Hum. Retroviruses 2016, 32, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Beig, J.; Orr, D.; Harrison, B.; Gane, E. Hepatitis C Virus Eradication with New Interferon-Free Treatment Improves Metabolic Profile in Hepatitis C Virus-Related Liver Transplant Recipients. Liver Transpl. 2018, 24, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.R.; Velosa, J.; Serejo, F. Lipids, glucose and iron metabolic alterations in chronic hepatitis C after viral eradication comparison of the new direct-acting antiviral agents with the old regimens. Scand J Gastroenterol. 2018, 53, 857–863. [Google Scholar] [CrossRef] [PubMed]
- El-Sagheer, G.; Soliman, E.; Ahmad, A.; Hamdy, L. Study of changes in lipid profile and insulin resistance in Egyptian patients with chronic hepatitis C genotype 4 in the era of DAAs. Libyan J. Med. 2018, 13, 1435124. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Colominas, E.; Batlle, M.; Monge-Escartin, I.; Duran, X.; Viu, A.; de Antonio-Cusco, M.; Grau, S.; Bessa, X.; Carrión, J.A. Impact of HCV cure with drug-acting antivirals in the use of concomitant medication and lipid profile: Follow-up data 2 years after the sustained virological response. Eur. J. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Doyle, M.A.; Galanakis, C.; Mulvihill, E.; Crawley, A.; Cooper, C.L. Hepatitis C Direct Acting Antivirals and Ribavirin Modify Lipid but not Glucose Parameters. Cells 2019, 8, 252. [Google Scholar] [CrossRef]
- Ichikawa, T.; Miyaaki, H.; Miuma, S.; Taura, N.; Motoyoshi, Y.; Akahoshi, H.; Nakamura, J.; Takahashi, Y.; Honda, T.; Yajima, H.; et al. Changes in serum LDL, PCSK9 and microRNA-122 in patients with chronic HCV infection receiving Daclatasvir/Asunaprevir. Biomed. Rep. 2019, 10, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Soroida, Y.; Sato, M.; Hikita, H.; Kobayashi, T.; Endo, M.; Sato, M.; Gotoh, H.; Iwai, T.; Tateishi, R.; et al. Eradication of hepatitis C virus is associated with the attenuation of steatosis as evaluated using a controlled attenuation parameter. Sci. Rep. 2018, 8, 7845. [Google Scholar] [CrossRef]
- Cheng, P.N.; Chen, J.Y.; Chiu, Y.C.; Chiu, H.C.; Tsai, L.M. Augmenting central arterial stiffness following eradication of HCV by direct acting antivirals in advanced fibrosis patients. Sci. Rep. 2019, 9, 1426. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Kalra, B.S.; Srivastava, S.; Chawla, S. Effect of sofosbuvir and daclatasvir on lipid profile, glycemic control and quality of life index in chronic hepatitis C, genotype 3 patients. Indian J. Gastroenterol. 2019, 38, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Maida, M.; Macaluso, F.S.; Barbara, M.; Licata, A.; Craxì, A.; Cammà, C. Hepatitis C Virus Infection Is Associated with Increased Cardiovascular Mortality: A Meta-Analysis of Observational Studies. Gastroenterology 2016, 150, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Casas-Deza, D.; Espina, S.; Martínez-Sapiña, A.; del Moral-Bergos, R.; Garcia-Sobreviela, M.P.; Lopez-Yus, M.; Calmarza, P.; Bernal-Monterde, V.; Arbones-Mainar, J.M. Triglyceride-rich lipoproteins and insulin resistance in patients with chronic hepatitis C receiving direct-acting antivirals. Atherosclerosis 2023, 375, 59–66. [Google Scholar] [CrossRef]
- Hashimoto, S.; Yatsuhashi, H.; Abiru, S.; Yamasaki, K.; Komori, A.; Nagaoka, S.; Saeki, A.; Uchida, S.; Bekki, S.; Kugiyama, Y.; et al. Rapid Increase in Serum Low-Density Lipoprotein Cholesterol Concentration during Hepatitis C Interferon-Free Treatment. PLoS ONE 2016, 11, e0163644. [Google Scholar] [CrossRef]
- Corey, K.E.; Kane, E.; Munroe, C.; Barlow, L.L.; Zheng, H.; Chung, R.T. Hepatitis C Virus Infection and Its Clearance Alter Circulating Lipids: Implications for Long-Term Follow-Up. 2009. Available online: https://aasldpubs.onlinelibrary.wiley.com/doi/10.1002/hep.23219 (accessed on 13 January 2024).
- Butt, A.A.; Yan, P.; Shuaib, A.; Abou-Samra, A.B.; Shaikh, O.S.; Freiberg, M.S. Direct-Acting Antiviral Therapy for HCV Infection Is Associated with a Reduced Risk of Cardiovascular Disease Events. Gastroenterology 2019, 156, 987–996. [Google Scholar] [CrossRef]
- Mahale, P.; Engels, E.A.; Li, R.; Torres, H.A.; Hwang, L.Y.; Brown, E.L.; Kramer, J.R. The effect of sustained virological response on the risk of extrahepatic manifestations of hepatitis C virus infection. Gut 2018, 67, 553. [Google Scholar] [CrossRef]
- Daskalopoulou, S.S.; Delaney, J.A.C.; Filion, K.B.; Brophy, J.M.; Mayo, N.E.; Suissa, S. Discontinuation of Statin Therapy Following an Acute Myocardial Infarction: A Population-Based Study. Available online: https://academic.oup.com/eurheartj/article/29/17/2083/578019 (accessed on 24 January 2024).
- Petta, S.; Adinolfi, L.E.; Fracanzani, A.L.; Rini, F.; Caldarella, R.; Calvaruso, V.; Cammà, C.; Ciacco, M.; Di-Marco, V.; Grimaudo, S.; et al. Hepatitis C virus eradication by direct-acting antiviral agents improves carotid atherosclerosis in patients with severe liver fibrosis. J. Hepatol. 2018, 69, 18–24. [Google Scholar] [CrossRef]
- A Systematic Review and Meta-Analysis of the Therapeutic Equivalence of Statins. Available online: www.hiv-druginteractions.org (accessed on 25 January 2024).


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pascual-Oliver, A.; Casas-Deza, D.; Yagüe-Caballero, C.; Arbones-Mainar, J.M.; Bernal-Monterde, V. Lipid Profile and Cardiovascular Risk Modification after Hepatitis C Virus Eradication. Pathogens 2024, 13, 278. https://doi.org/10.3390/pathogens13040278
Pascual-Oliver A, Casas-Deza D, Yagüe-Caballero C, Arbones-Mainar JM, Bernal-Monterde V. Lipid Profile and Cardiovascular Risk Modification after Hepatitis C Virus Eradication. Pathogens. 2024; 13(4):278. https://doi.org/10.3390/pathogens13040278
Chicago/Turabian StylePascual-Oliver, Andrea, Diego Casas-Deza, Carmen Yagüe-Caballero, Jose M. Arbones-Mainar, and Vanesa Bernal-Monterde. 2024. "Lipid Profile and Cardiovascular Risk Modification after Hepatitis C Virus Eradication" Pathogens 13, no. 4: 278. https://doi.org/10.3390/pathogens13040278
APA StylePascual-Oliver, A., Casas-Deza, D., Yagüe-Caballero, C., Arbones-Mainar, J. M., & Bernal-Monterde, V. (2024). Lipid Profile and Cardiovascular Risk Modification after Hepatitis C Virus Eradication. Pathogens, 13(4), 278. https://doi.org/10.3390/pathogens13040278