Case Report: Shift from Aggressive Periodontitis to Feline Chronic Gingivostomatitis Is Linked to Increased Microbial Diversity



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
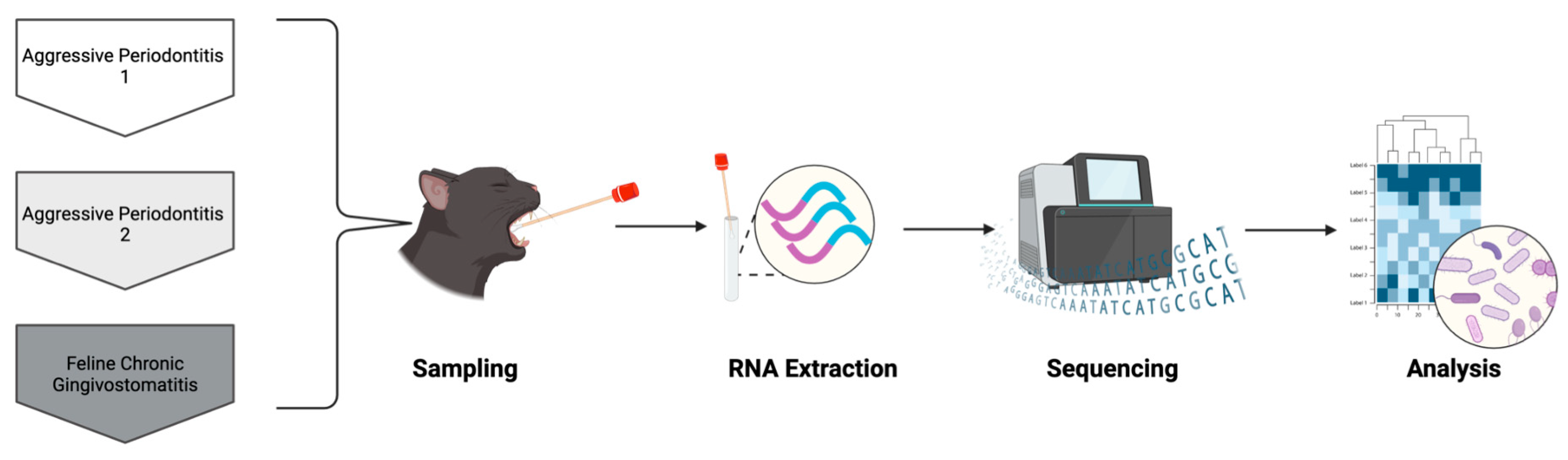
2. Case Description
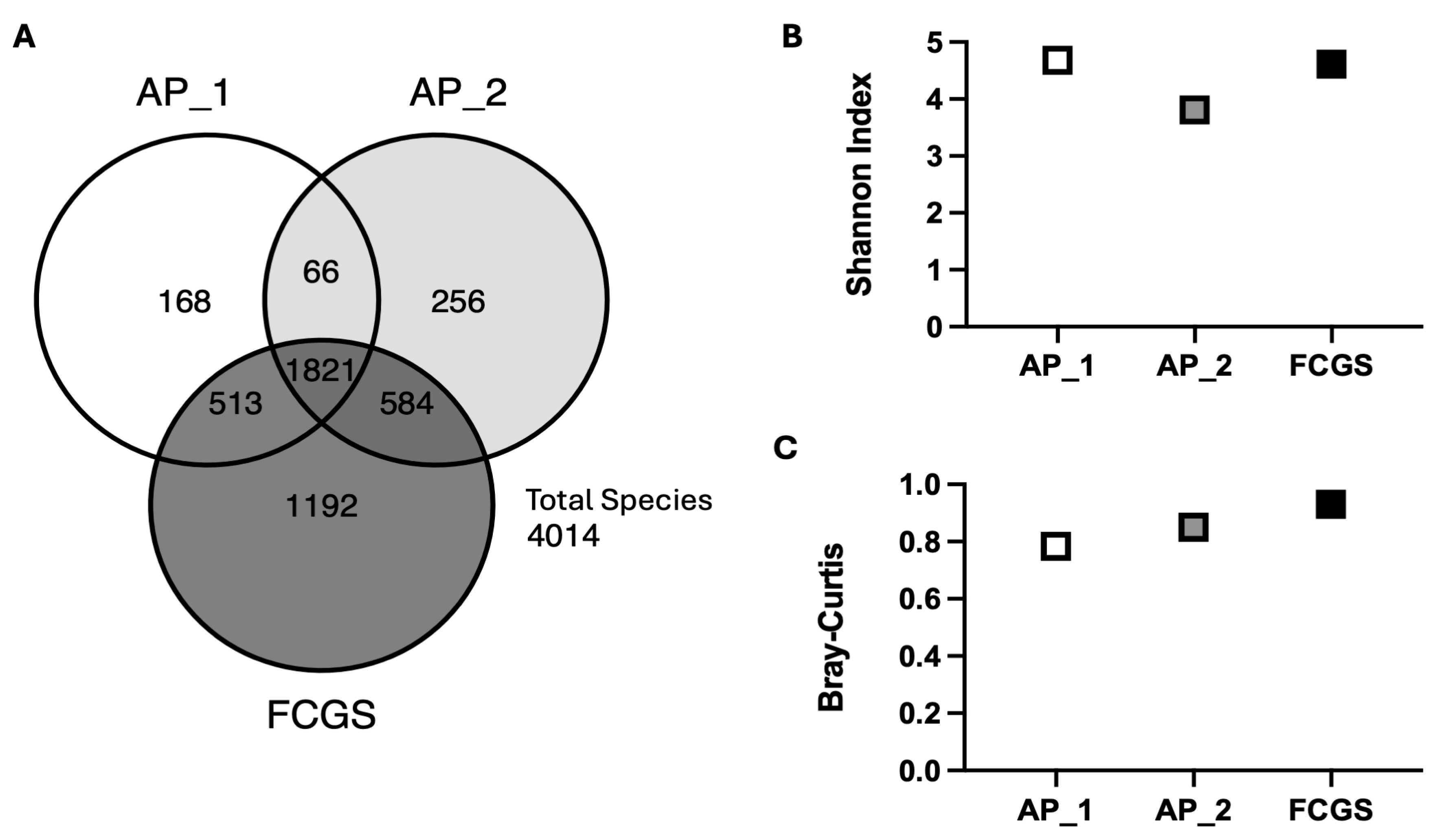
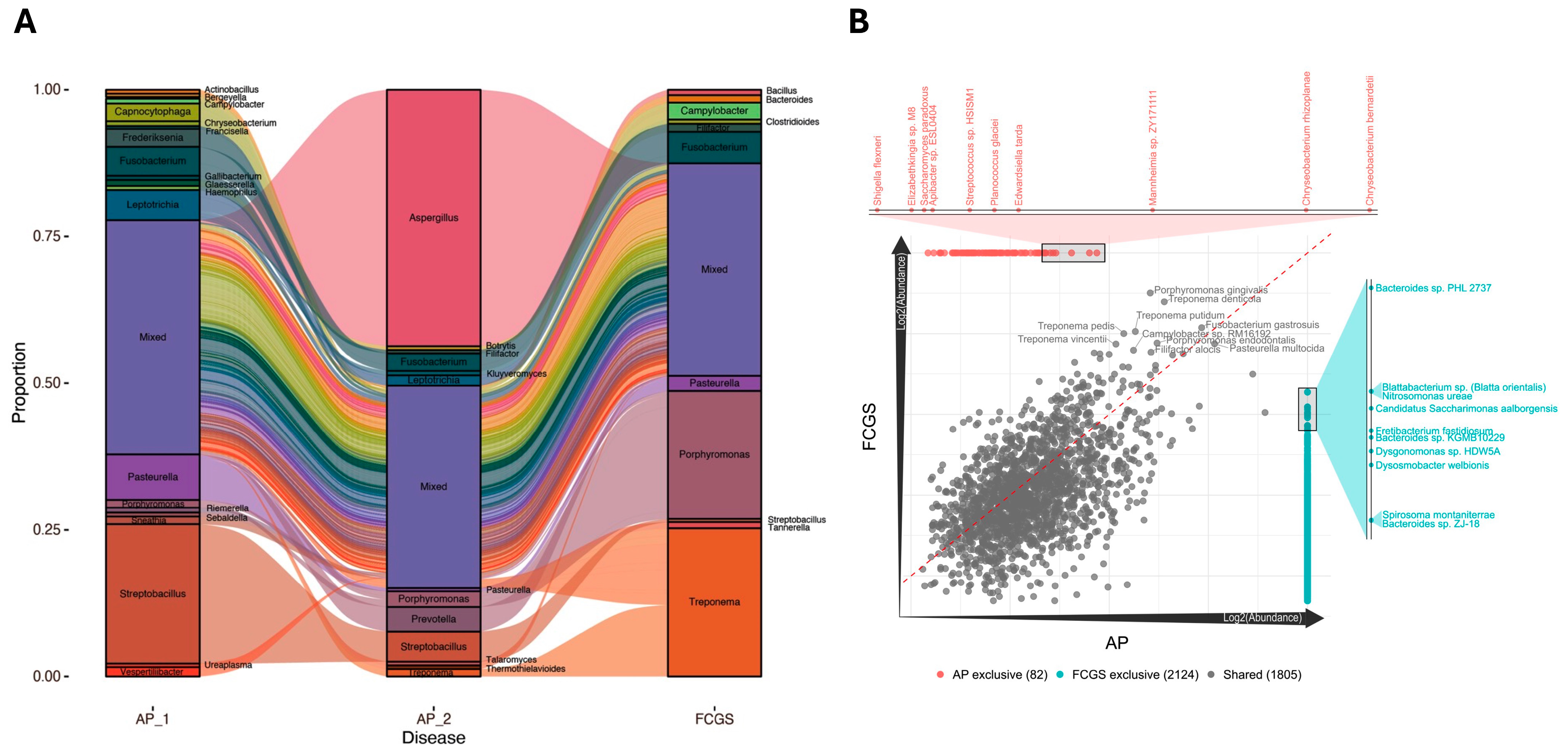
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Soltero-Rivera, M.; Vapniarsky, N.; Rivas, I.L.; Arzi, B. Clinical, radiographic and histopathologic features of early-onset gingivitis and periodontitis in cats (1997–2022). J. Feline Med. Surg. 2023, 25, 1098612X221148577. [Google Scholar] [CrossRef] [PubMed]
- Soltero-Rivera, M.; Goldschmidt, S.; Arzi, B. Feline chronic gingivostomatitis current concepts in clinical management. J. Feline Med. Surg. 2023, 25, 1098612X231186834. [Google Scholar] [CrossRef] [PubMed]
- Dolieslager, S.M.; Lappin, D.F.; Bennett, D.; Graham, L.; Johnston, N.; Riggio, M.P. The influence of oral bacteria on tissue levels of Toll-like receptor and cytokine mRNAs in feline chronic gingivostomatitis and oral health. Vet. Immunol. Immunopathol. 2013, 151, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Dolieslager, S.M.; Riggio, M.P.; Lennon, A.; Lappin, D.F.; Johnston, N.; Taylor, D.; Bennett, D. Identification of bacteria associated with feline chronic gingivostomatitis using culture-dependent and culture-independent methods. Vet. Microbiol. 2011, 148, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Older, C.E.; Gomes, M.O.S.; Hoffmann, A.R.; Policano, M.D.; Reis, C.; Carregaro, A.B.; Ambrosio, C.E.; Carregaro, V.M.L. Influence of the FIV Status and Chronic Gingivitis on Feline Oral Microbiota. Pathogens 2020, 9, 383. [Google Scholar] [CrossRef] [PubMed]
- Peralta, S.; Grenier, J.K.; Webb, S.M.; Miller, A.D.; Miranda, I.C.; Parker, J.S.L. Transcriptomic signatures of feline chronic gingivostomatitis are influenced by upregulated IL6. Sci. Rep. 2023, 13, 13437. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.; Tutt, C. Periodontal disease in cats: Back to basics–with an eye on the future. J. Feline Med. Surg. 2015, 17, 45–65. [Google Scholar] [CrossRef]
- Shaw, C.A.; Soltero-Rivera, M.; Profeta, R.; Weimer, B.C. Case Report: Inflammation-Driven Species-Level Shifts in the Oral Microbiome of Refractory Feline Chronic Gingivostomatitis. Bacteria 2025, 4, 1. [Google Scholar] [CrossRef]
- Soltero-Rivera, M.; Shaw, C.; Arzi, B.; Lommer, M.; Weimer, B.C. Feline Chronic Gingivostomatitis Diagnosis and Treatment through Transcriptomic Insights. Pathogens 2024, 13, 192. [Google Scholar] [CrossRef] [PubMed]
- Winer, J.N.; Arzi, B.; Verstraete, F.J. Therapeutic Management of Feline Chronic Gingivostomatitis: A Systematic Review of the Literature. Front. Vet. Sci. 2016, 3, 54. [Google Scholar] [CrossRef]
- Davis, E.M. Gene Sequence Analyses of the Healthy Oral Microbiome in Humans and Companion Animals. J. Vet. Dent. 2016, 33, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Duran-Pinedo, A.E.; Frias-Lopez, J. Beyond microbial community composition: Functional activities of the oral microbiome in health and disease. Microbes Infect. 2015, 17, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.M.; Weese, J.S. Oral Microbiome in Dogs and Cats: Dysbiosis and the Utility of Antimicrobial Therapy in the Treatment of Periodontal Disease. Vet. Clin. N. Am. Small Anim. Pract. 2022, 52, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.; Croft, J.; O’Flynn, C.; Deusch, O.; Colyer, A.; Allsopp, J.; Milella, L.; Davis, I.J. A Pyrosequencing Investigation of Differences in the Feline Subgingival Microbiota in Health, Gingivitis and Mild Periodontitis. PLoS ONE 2015, 10, e0136986. [Google Scholar] [CrossRef]
- Rodrigues, M.X.; Bicalho, R.C.; Fiani, N.; Lima, S.F.; Peralta, S. The subgingival microbial community of feline periodontitis and gingivostomatitis: Characterization and comparison between diseased and healthy cats. Sci. Rep. 2019, 9, 12340. [Google Scholar] [CrossRef] [PubMed]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The oral microbiota: Dynamic communities and host interactions. Nat. Rev. Microbiol. 2018, 16, 745–759. [Google Scholar] [CrossRef]
- Moreno, C.M.; Boeree, E.; Freitas, C.M.T.; Weber, K.S. Immunomodulatory role of oral microbiota in inflammatory diseases and allergic conditions. Front. Allergy 2023, 4, 1067483. [Google Scholar] [CrossRef] [PubMed]
- Barbour, A.; Elebyary, O.; Fine, N.; Oveisi, M.; Glogauer, M. Metabolites of the oral microbiome: Important mediators of multikingdom interactions. FEMS Microbiol. Rev. 2022, 46, fuab039. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; He, F.; Si, M.; Sun, P.; Chen, Q. Effects of Antibiotic Use on Saliva Antibody Content and Oral Microbiota in Sprague Dawley Rats. Front. Cell Infect. Microbiol. 2022, 12, 721691. [Google Scholar] [CrossRef]
- Adler, C.J.; Malik, R.; Browne, G.V.; Norris, J.M. Diet may influence the oral microbiome composition in cats. Microbiome 2016, 4, 23. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Chung, S.W.; Auh, Q.S.; Hong, S.J.; Lee, Y.A.; Jung, J.; Lee, G.J.; Park, H.J.; Shin, S.I.; Hong, J.Y. Progress in Oral Microbiome Related to Oral and Systemic Diseases: An Update. Diagnostics 2021, 11, 1283. [Google Scholar] [CrossRef]
- Bui, F.Q.; Almeida-da-Silva, C.L.C.; Huynh, B.; Trinh, A.; Liu, J.; Woodward, J.; Asadi, H.; Ojcius, D.M. Association between periodontal pathogens and systemic disease. Biomed. J. 2019, 42, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Mysak, J.; Podzimek, S.; Sommerova, P.; Lyuya-Mi, Y.; Bartova, J.; Janatova, T.; Prochazkova, J.; Duskova, J. Porphyromonas gingivalis: Major periodontopathic pathogen overview. J. Immunol. Res. 2014, 2014, 476068. [Google Scholar] [CrossRef] [PubMed]
- Santonocito, S.; Giudice, A.; Polizzi, A.; Troiano, G.; Merlo, E.M.; Sclafani, R.; Grosso, G.; Isola, G. A Cross-Talk between Diet and the Oral Microbiome: Balance of Nutrition on Inflammation and Immune System’s Response during Periodontitis. Nutrients 2022, 14, 2426. [Google Scholar] [CrossRef] [PubMed]
- Sedghi, L.M.; Bacino, M.; Kapila, Y.L. Periodontal Disease: The Good, The Bad, and The Unknown. Front. Cell Infect. Microbiol. 2021, 11, 766944. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.H.; Seers, C.A.; Dashper, S.G.; Mitchell, H.L.; Pyke, J.S.; Meuric, V.; Slakeski, N.; Cleal, S.M.; Chambers, J.L.; McConville, M.J.; et al. Porphyromonas gingivalis and Treponema denticola exhibit metabolic symbioses. PLoS Pathog. 2014, 10, e1003955. [Google Scholar] [CrossRef] [PubMed]
- Meuric, V.; Martin, B.; Guyodo, H.; Rouillon, A.; Tamanai-Shacoori, Z.; Barloy-Hubler, F.; Bonnaure-Mallet, M. Treponema denticola improves adhesive capacities of Porphyromonas gingivalis. Mol. Oral. Microbiol. 2013, 28, 40–53. [Google Scholar] [CrossRef]
- Nibali, L. Aggressive Periodontitis: Microbes and host response, who to blame? Virulence 2015, 6, 223–228. [Google Scholar] [CrossRef]
- Kong, N.; Ng, W.; Lee, V.; Kelly, L.; Weimer, B. Production and Analysis of High Molecular Weight Genomic DNA for NGS Pipelines Using Agilent DNA Extraction Kit (p/n 200600); Agilent Technologies: Santa Clara, CA, USA, 2013; Volume 10. [Google Scholar]
- Basbas, C.; Garzon, A.; Schlesener, C.; van Heule, M.; Profeta, R.; Weimer, B.C.; Silva-Del-Rio, N.; Byrne, B.A.; Karle, B.; Aly, S.S.; et al. Unveiling the microbiome during post-partum uterine infection: A deep shotgun sequencing approach to characterize the dairy cow uterine microbiome. Anim. Microbiome 2023, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Garzon, A.; Basbas, C.; Schlesener, C.; Silva-Del-Rio, N.; Karle, B.M.; Lima, F.S.; Weimer, B.C.; Pereira, R.V. WGS of intrauterine E. coli from cows with early postpartum uterine infection reveals a non-uterine specific genotype and virulence factors. mBio 2024, 15, e0102724. [Google Scholar] [CrossRef] [PubMed]
- Beck, K.L.; Haiminen, N.; Chambliss, D.; Edlund, S.; Kunitomi, M.; Huang, B.C.; Kong, N.; Ganesan, B.; Baker, R.; Markwell, P.; et al. Monitoring the microbiome for food safety and quality using deep shotgun sequencing. npj Sci. Food 2021, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Huynh, W.; Chalil, R.; Smith, K.W.; Raphenya, A.R.; Wlodarski, M.A.; Edalatmand, A.; Petkau, A.; Syed, S.A.; Tsang, K.K.; et al. CARD 2023: Expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2023, 51, D690–D699. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, L.; Sun, L.; Yu, J.; Jin, Q. VFDB 2008 release: An enhanced web-based resource for comparative pathogenomics. Nucleic Acids Res. 2008, 36, D539–D542. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Faller, L.L.; Klitgord, N.; Mazumdar, V.; Ghodsi, M.; Sommer, D.D.; Gibbons, T.R.; Treangen, T.J.; Chang, Y.C.; Li, S.; et al. Deep sequencing of the oral microbiome reveals signatures of periodontal disease. PLoS ONE 2012, 7, e37919. [Google Scholar] [CrossRef] [PubMed]
- Lenartova, M.; Tesinska, B.; Janatova, T.; Hrebicek, O.; Mysak, J.; Janata, J.; Najmanova, L. The Oral Microbiome in Periodontal Health. Front. Cell Infect. Microbiol. 2021, 11, 629723. [Google Scholar] [CrossRef] [PubMed]
- Matsha, T.E.; Prince, Y.; Davids, S.; Chikte, U.; Erasmus, R.T.; Kengne, A.P.; Davison, G.M. Oral Microbiome Signatures in Diabetes Mellitus and Periodontal Disease. J. Dent. Res. 2020, 99, 658–665. [Google Scholar] [CrossRef]
- Munoz Navarro, C.; Del Carmen Sanchez Beltran, M.; Arriagada Vargas, C.; Batalla Vazquez, P.; Diniz Freitas, M.; Limeres Posse, J.; Diz Dios, P.; Garcia Mato, E. Analysis of the Oral Microbiome in a Patient with Cardiofaciocutaneous Syndrome and Severe Periodontal Disease: Impact of Systemic Antibiotic Therapy. Antibiotics 2022, 11, 1754. [Google Scholar] [CrossRef]
- Stone, A.E.S. Feline oral inflammation: Diagnosis and options for treatment. Companion Anim. 2024, 29, 112–118. [Google Scholar] [CrossRef]
- Antezack, A.; Etchecopar-Etchart, D.; La Scola, B.; Monnet-Corti, V. New putative periodontopathogens and periodontal health-associated species: A systematic review and meta-analysis. J. Periodontal Res. 2023, 58, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, R.; Badran, Z.; Boghossian, A.; Alharbi, A.M.; Alfahemi, H.; Khan, N.A. The increasing importance of the oral microbiome in periodontal health and disease. Future Sci. OA 2023, 9, FSO856. [Google Scholar] [CrossRef] [PubMed]
- Sudhakara, P.; Gupta, A.; Bhardwaj, A.; Wilson, A. Oral Dysbiotic Communities and Their Implications in Systemic Diseases. Dent. J. 2018, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, R.; Asopa, S.J.; Joseph, M.D.; Singh, B.; Rajguru, J.P.; Saidath, K.; Sharma, U. Red complex: Polymicrobial conglomerate in oral flora: A review. J. Fam. Med. Prim. Care 2019, 8, 3480–3486. [Google Scholar] [CrossRef]
- Older, C.E.; Diesel, A.B.; Lawhon, S.D.; Queiroz, C.R.R.; Henker, L.C.; Rodrigues Hoffmann, A. The feline cutaneous and oral microbiota are influenced by breed and environment. PLoS ONE 2019, 14, e0220463. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi-Kuroda, Y.; Kikuchi, Y.; Kokubu, E.; Ishihara, K. Porphyromonas gingivalis diffusible signaling molecules enhance Fusobacterium nucleatum biofilm formation via gene expression modulation. J. Oral. Microbiol. 2023, 15, 2165001. [Google Scholar] [CrossRef]
- Cools, F.; Torfs, E.; Vanhoutte, B.; de Macedo, M.B.; Bonofiglio, L.; Mollerach, M.; Maes, L.; Caljon, G.; Delputte, P.; Cappoen, D.; et al. Streptococcus pneumoniae galU gene mutation has a direct effect on biofilm growth, adherence and phagocytosis in vitro and pathogenicity in vivo. Pathog. Dis. 2018, 76, fty069. [Google Scholar] [CrossRef] [PubMed]
- Lasserre, J.F.; Brecx, M.C.; Toma, S. Oral Microbes, Biofilms and Their Role in Periodontal and Peri-Implant Diseases. Materials 2018, 11, 1802. [Google Scholar] [CrossRef]
- Shi, M.; Wei, Y.; Hu, W.; Nie, Y.; Wu, X.; Lu, R. The Subgingival Microbiome of Periodontal Pockets With Different Probing Depths in Chronic and Aggressive Periodontitis: A Pilot Study. Front. Cell Infect. Microbiol. 2018, 8, 124. [Google Scholar] [CrossRef]
- Johnston, W.; Rosier, B.T.; Artacho, A.; Paterson, M.; Piela, K.; Delaney, C.; Brown, J.L.; Ramage, G.; Mira, A.; Culshaw, S. Mechanical biofilm disruption causes microbial and immunological shifts in periodontitis patients. Sci. Rep. 2021, 11, 9796. [Google Scholar] [CrossRef] [PubMed]
- Ingendoh-Tsakmakidis, A.; Mikolai, C.; Winkel, A.; Szafranski, S.P.; Falk, C.S.; Rossi, A.; Walles, H.; Stiesch, M. Commensal and pathogenic biofilms differently modulate peri-implant oral mucosa in an organotypic model. Cell Microbiol. 2019, 21, e13078. [Google Scholar] [CrossRef] [PubMed]
- Mark Welch, J.L.; Rossetti, B.J.; Rieken, C.W.; Dewhirst, F.E.; Borisy, G.G. Biogeography of a human oral microbiome at the micron scale. Proc. Natl. Acad. Sci. USA 2016, 113, E791–E800. [Google Scholar] [CrossRef] [PubMed]
- Bacali, C.; Vulturar, R.; Buduru, S.; Cozma, A.; Fodor, A.; Chis, A.; Lucaciu, O.; Damian, L.; Moldovan, M.L. Oral Microbiome: Getting to Know and Befriend Neighbors, a Biological Approach. Biomedicines 2022, 10, 671. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, S.-I.; Minamino, T. Chapter 6—Flagella. In Molecular Medical Microbiology, 3rd ed.; Tang, Y.-W., Hindiyeh, M.Y., Liu, D., Sails, A., Spearman, P., Zhang, J.-R., Eds.; Academic Press: Cambridge, MA, USA, 2024; pp. 97–126. [Google Scholar] [CrossRef]
- Scannapieco, F.A.; Kornman, K.S.; Coykendall, A.L. Observation of fimbriae and flagella in dispersed subgingival dental plaque and fresh bacterial isolates from periodontal disease. J. Periodontal Res. 1983, 18, 620–633. [Google Scholar] [CrossRef]
- Goetting-Minesky, M.P.; Godovikova, V.; Fenno, J.C. Approaches to Understanding Mechanisms of Dentilisin Protease Complex Expression in Treponema denticola. Front. Cell Infect. Microbiol. 2021, 11, 668287. [Google Scholar] [CrossRef] [PubMed]
- Lux, R.; Miller, J.N.; Park, N.H.; Shi, W. Motility and chemotaxis in tissue penetration of oral epithelial cell layers by Treponema denticola. Infect. Immun. 2001, 69, 6276–6283. [Google Scholar] [CrossRef] [PubMed]
- Ruby, J.; Martin, M.; Passineau, M.J.; Godovikova, V.; Fenno, J.C.; Wu, H. Activation of the Innate Immune System by Treponema denticola Periplasmic Flagella through Toll-Like Receptor 2. Infect. Immun. 2018, 86, e00573-17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Szymanski, C.M.; Baylink, A. Bacterial chemotaxis in human diseases. Trends Microbiol. 2023, 31, 453–467. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaw, C.A.; Soltero-Rivera, M.; Profeta, R.; Weimer, B.C. Case Report: Shift from Aggressive Periodontitis to Feline Chronic Gingivostomatitis Is Linked to Increased Microbial Diversity. Pathogens 2025, 14, 228. https://doi.org/10.3390/pathogens14030228
Shaw CA, Soltero-Rivera M, Profeta R, Weimer BC. Case Report: Shift from Aggressive Periodontitis to Feline Chronic Gingivostomatitis Is Linked to Increased Microbial Diversity. Pathogens. 2025; 14(3):228. https://doi.org/10.3390/pathogens14030228
Chicago/Turabian StyleShaw, Claire A., Maria Soltero-Rivera, Rodrigo Profeta, and Bart C. Weimer. 2025. "Case Report: Shift from Aggressive Periodontitis to Feline Chronic Gingivostomatitis Is Linked to Increased Microbial Diversity" Pathogens 14, no. 3: 228. https://doi.org/10.3390/pathogens14030228
APA StyleShaw, C. A., Soltero-Rivera, M., Profeta, R., & Weimer, B. C. (2025). Case Report: Shift from Aggressive Periodontitis to Feline Chronic Gingivostomatitis Is Linked to Increased Microbial Diversity. Pathogens, 14(3), 228. https://doi.org/10.3390/pathogens14030228