
Recombinant Microneme Proteins MIC1 and MIC4 from Toxoplasma gondii Cause Cytotoxic Effects in the Human Jurkat T-Lymphocyte Cell Line


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Jurkat Cell Cultures
2.2. Recombinant Proteins
2.3. Binding Assay
2.4. Evaluation of Jurkat Cells Viability After Stimulus with rMIC1 or rMIC4
2.5. Apoptosis Detection
2.6. Caspase Activity Evaluation
2.7. Assessment of the Mitochondrial Membrane Potential
2.8. Chromatin Fragmentation Analysis
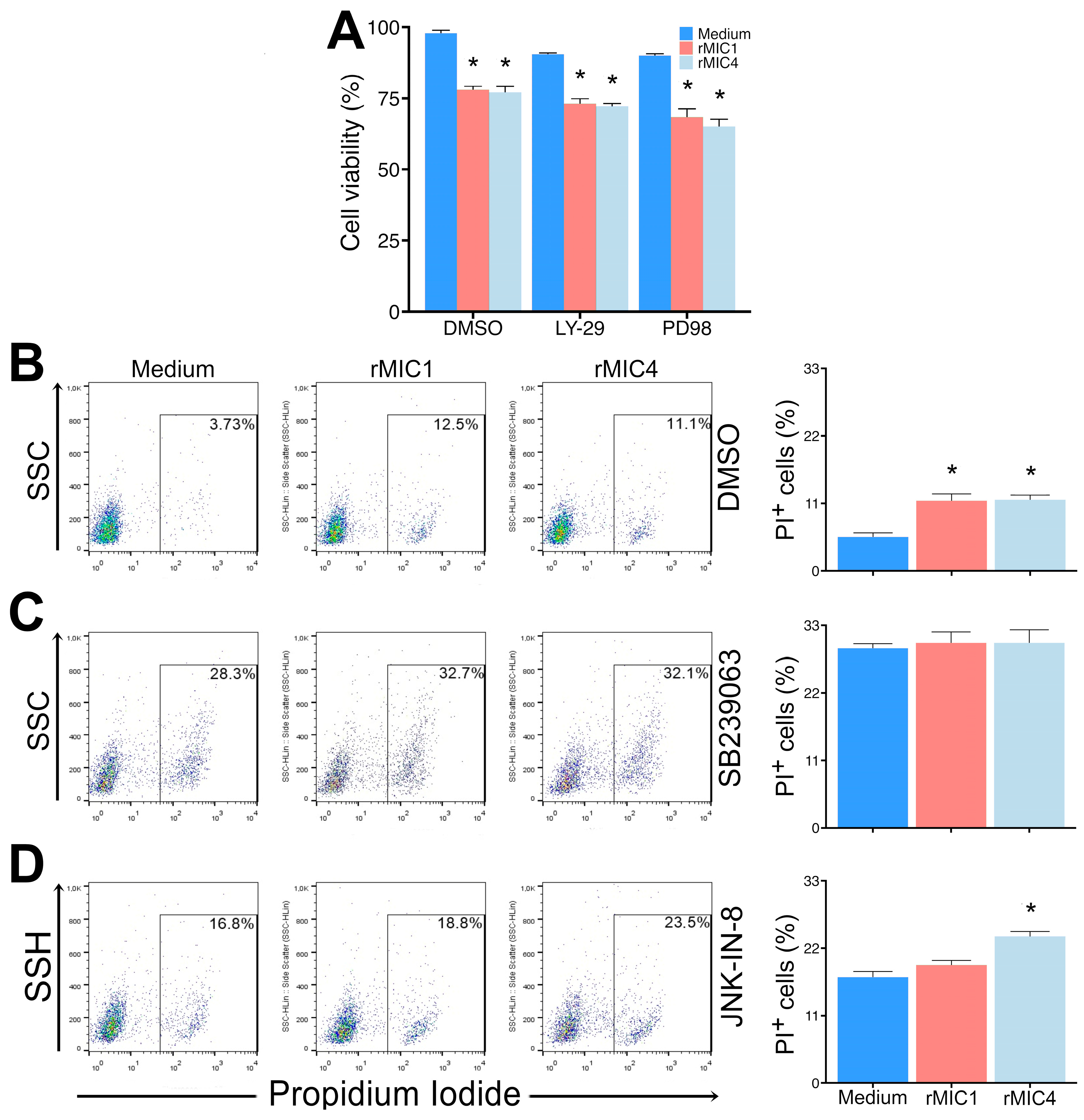
2.9. Identification of Cell Signaling Pathways Used by MICs to Cause Cell Death
2.10. Detection of ROS
2.11. Statistical Analysis
3. Results
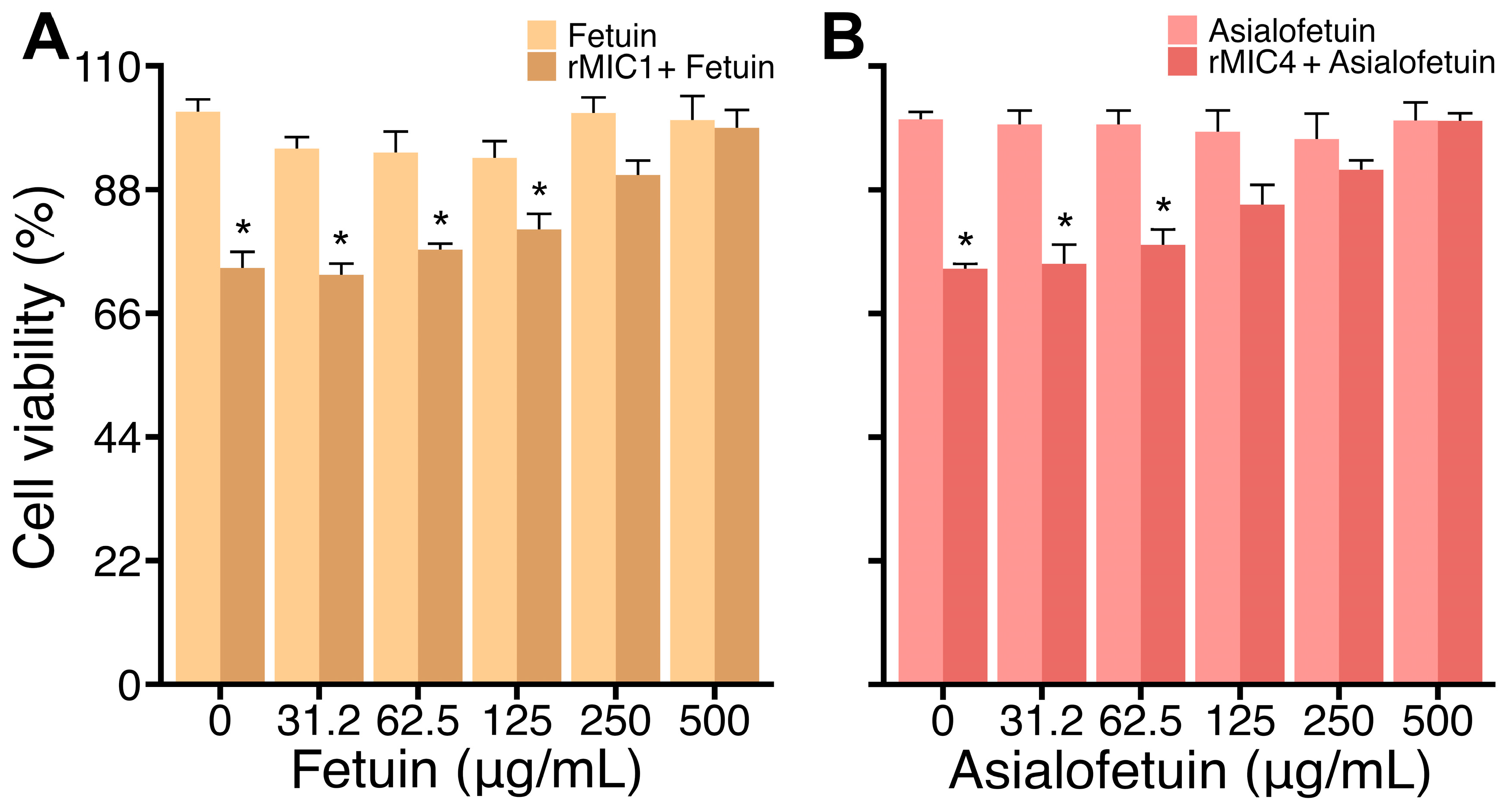
3.1. rMIC1 and rMIC4 Interact with Glycans on Jurkat Cells
3.2. rMIC1 and rMIC4 Induce Time- and Dose-Dependent Loss of Viability and Growth Inhibition in Jurkat Cells
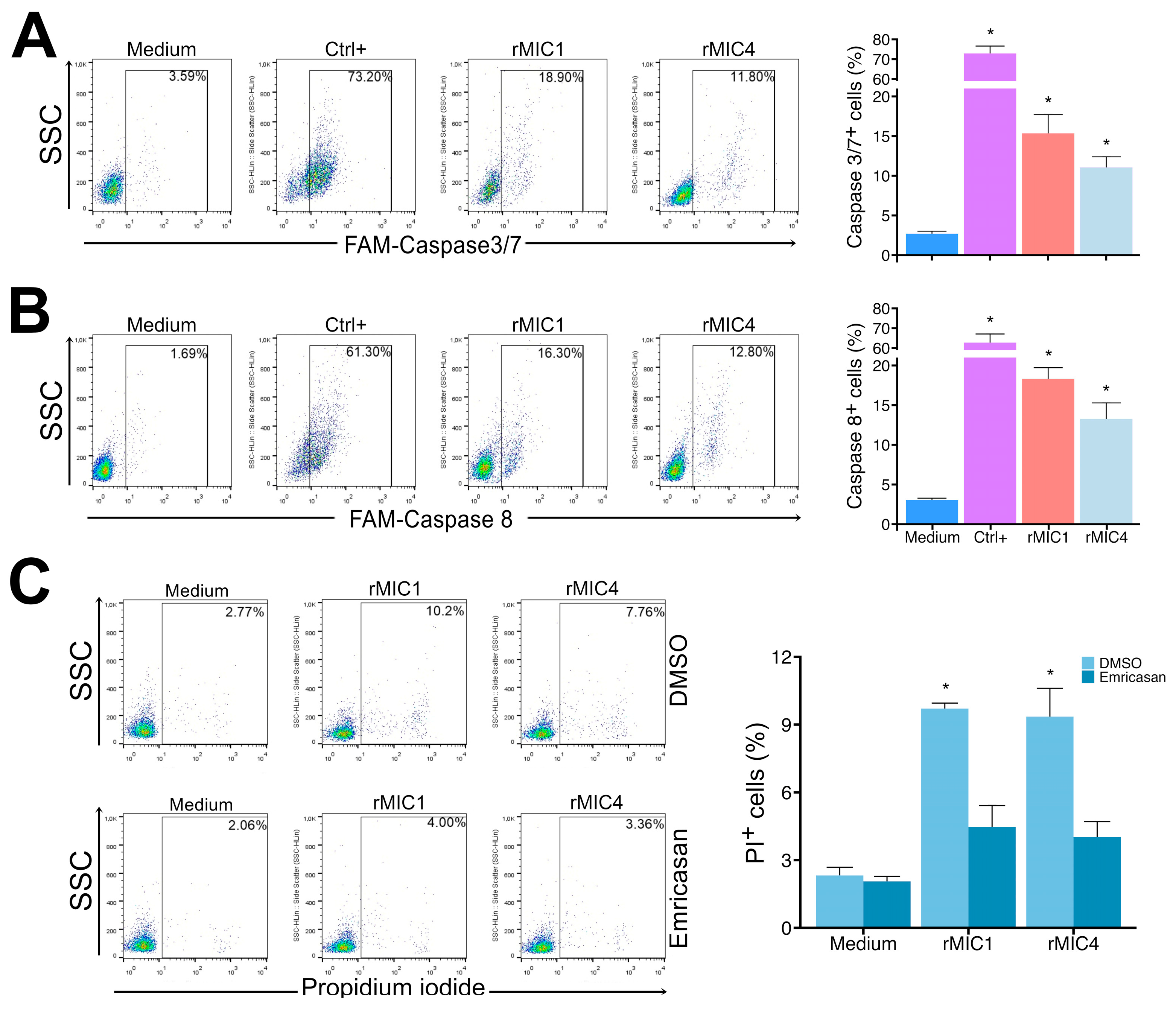
3.3. rMIC1 and rMIC4 Induce Apoptosis in Jurkat Cells
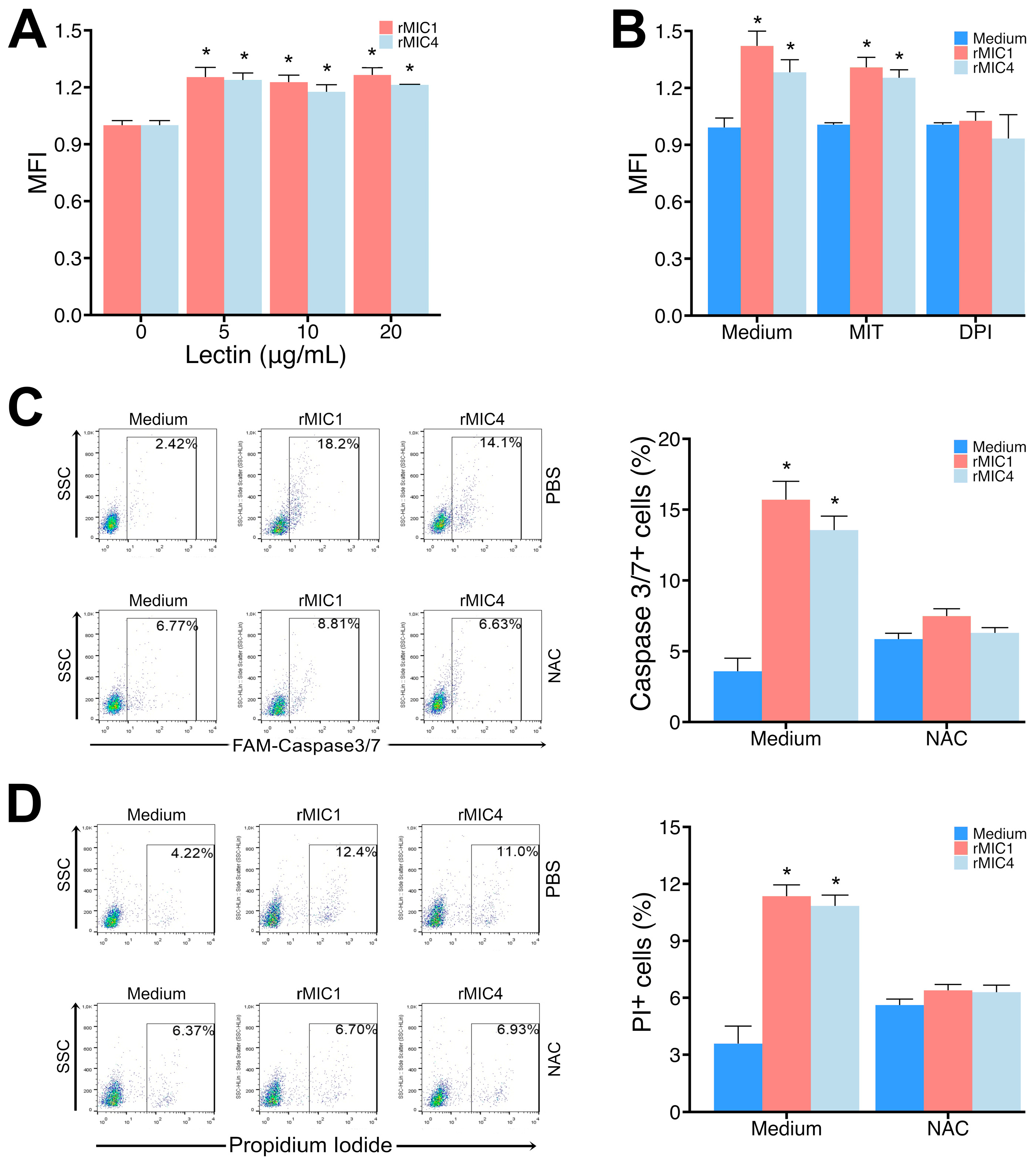
3.4. Release of ROS from the NADPH Oxidase Complex Is Responsible for Apoptosis of Jurkat Cells Induced by rMIC1 or rMIC4
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Akins, G.K.H.; Furtado, J.M.; Smith, J.R. Diseases Caused by and Behaviors Associated with Toxoplasma gondii Infection. Pathogens 2024, 13, 968. [Google Scholar] [CrossRef] [PubMed]
- Dunay, I.R.; Gajurel, K.; Dhakal, R.; Liesenfeld, O.; Montoya, J.G. Treatment of Toxoplasmosis: Historical Perspective, Animal Models, and Current Clinical Practice. Clin. Microbiol. Rev. 2018, 31, e00057-17. [Google Scholar] [CrossRef]
- Torgerson, P.R.; Mastroiacovo, P. The Global Burden of Congenital Toxoplasmosis: A Systematic Review. Bull. World Health Organ. 2013, 91, 501–508. [Google Scholar] [CrossRef]
- Carruthers, V.B.; Sibley, L.D. Sequential Protein Secretion Front Three Distinct Organelles of Toxoplasma gondii Accompanies Invasion of Human Fibroblasts. Eur. J. Cell Biol. 1997, 73, 114–123. [Google Scholar]
- Portes, J.; Barrias, E.; Travassos, R.; Attias, M.; de Souza, W. Toxoplasma gondii Mechanisms of Entry into Host Cells. Front. Cell Infect. Microbiol. 2020, 10, 294. [Google Scholar] [CrossRef]
- Ben Chaabene, R.; Lentini, G.; Soldati-Favre, D. Biogenesis and Discharge of the Rhoptries: Key Organelles for Entry and Hijack of Host Cells by the Apicomplexa. Mol. Microbiol. 2021, 115, 453–465. [Google Scholar] [CrossRef]
- Liu, Q.; Li, F.C.; Zhou, C.X.; Zhu, X.Q. Research Advances in Interactions Related to Toxoplasma gondii Microneme Proteins. Exp. Parasitol. 2017, 176, 89–98. [Google Scholar] [PubMed]
- Zhu, J.; Wang, Y.; Cao, Y.; Shen, J.; Yu, L. Diverse Roles of TgMIC1/4/6 in the Toxoplasma Infection. Front. Microbiol. 2021, 12, 665606. [Google Scholar] [CrossRef]
- Reiss, M.; Viebig, N.; Brecht, S.; Fourmaux, M.N.; Soete, M.; Di Cristina, M.; Dubremetz, J.F.; Soldati, D. Identification and Characterization of an Escorter for Two Secretory Adhesins in Toxoplasma gondii. J. Cell Biol. 2001, 153, 563–578. [Google Scholar] [CrossRef]
- Friedrich, N.; Santos, J.M.; Liu, Y.; Palma, A.S.; Leon, E.; Saouros, S.; Kiso, M.; Blackman, M.J.; Matthews, S.; Feizi, T.; et al. Members of a Novel Protein Family Containing Microneme Adhesive Repeat Domains Act as Sialic Acid-Binding Lectins during Host Cell Invasion by Apicomplexan Parasites. J. Biol. Chem. 2010, 285, 2064–2076. [Google Scholar] [CrossRef]
- Garnett, J.A.; Liu, Y.; Leon, E.; Allman, S.A.; Friedrich, N.; Saouros, S.; Curry, S.; Soldati-Favre, D.; Davis, B.G.; Feizi, T.; et al. Detailed Insights from Microarray and Crystallographic Studies into Carbohydrate Recognition by Microneme Protein 1 (MIC1) of Toxoplasma gondii. Protein Sci. 2009, 18, 1935–1947. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, E.V.; Pereira, S.R.; Faça, V.M.; Coelho-Castelo, A.A.M.; Mineo, J.R.; Roque-Barreira, M.C.; Greene, L.J.; Panunto-Castelo, A. Toxoplasma gondii Micronemal Protein MIC1 Is a Lactose-Binding Lectin. Glycobiology 2001, 11, 541–547. [Google Scholar] [CrossRef]
- Marchant, J.; Cowper, B.; Liu, Y.; Lai, L.; Pinzan, C.; Marq, J.B.; Friedrich, N.; Sawmynaden, K.; Liew, L.; Chai, W.; et al. Galactose Recognition by the Apicomplexan Parasite Toxoplasma gondii. J. Biol. Chem. 2012, 287, 16720–16733. [Google Scholar] [CrossRef] [PubMed]
- Sardinha-Silva, A.; Mendonça-Natividade, F.C.; Pinzan, C.F.; Lopes, C.D.; Costa, D.L.; Jacot, D.; Fernandes, F.F.; Zorzetto-Fernandes, A.L.V.; Gay, N.J.; Sher, A.; et al. The Lectin-Specific Activity of Toxoplasma gondii Microneme Proteins 1 and 4 Binds Toll-like Receptor 2 and 4 N-Glycans to Regulate Innate Immune Priming. PLoS Pathog. 2019, 15, e1007871. [Google Scholar] [CrossRef]
- Mendonça-Natividade, F.C.; Lopes, C.D.; Ricci-Azevedo, R.; Sardinha-Silva, A.; Pinzan, C.F.; Alegre-Maller, A.C.P.; Nohara, L.L.; Carneiro, A.B.; Panunto-Castelo, A.; Almeida, I.C.; et al. Receptor Heterodimerization and Co-Receptor Engagement in TLR2 Activation Induced by MIC1 and MIC4 from Toxoplasma gondii. Int. J. Mol. Sci. 2019, 20, 5001. [Google Scholar] [CrossRef]
- Ricci-Azevedo, R.; Mendonça-Natividade, F.C.; Santana, A.C.; Alcoforado Diniz, J.; Roque-Barreira, M.C. Microneme Proteins 1 and 4 from Toxoplasma gondii Induce IL-10 Production by Macrophages through TLR4 Dndocytosis. Front. Immunol. 2021, 12, 655371. [Google Scholar] [CrossRef] [PubMed]
- Yarovinsky, F. Toll-like Receptors and Their Role in Host Resistance to Toxoplasma gondii. Immunol. Lett. 2008, 119, 17–21. [Google Scholar] [CrossRef]
- Yarovinsky, F. Innate Immunity to Toxoplasma gondii Infection. Nat. Rev. Immunol. 2014, 14, 109–121. [Google Scholar] [CrossRef]
- Ihara, F.; Yamamoto, M. The Role of IFN-γ-Mediated Host Immune Responses in Monitoring and the Elimination of Toxoplasma gondii Infection. Int. Immunol. 2024, 36, 199–210. [Google Scholar] [CrossRef]
- Lourenço, E.V.; Bernardes, E.S.; Silva, N.M.; Mineo, J.R.; Panunto-Castelo, A.; Roque-Barreira, M.C. Immunization with MIC1 and MIC4 Induces Protective Immunity against Toxoplasma gondii. Microbes Infect. 2006, 8, 1244–1251. [Google Scholar] [CrossRef]
- Pinzan, C.F.; Sardinha-Silva, A.; Almeida, F.; Lai, L.; Lopes, C.D.; Lourenço, E.V.; Panunto-Castelo, A.; Matthews, S.; Roque-Barreira, M.C. Vaccination with Recombinant Microneme Proteins Confers Protection against Experimental Toxoplasmosis in Mice. PLoS ONE 2015, 10, e0143087. [Google Scholar] [CrossRef]
- Taylor, M.E.; Drickamer, K.; Imberty, A.; van Kooyk, Y.; Schnaar, R.L.; Etzler, M.E.; Varki, A. Discovery and Classification of Glycan-Binding Proteins. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2022. [Google Scholar]
- Griffiths, G.D.; Leek, M.D.; Gee, D.J. The Toxic Plant Proteins Ricin and Abrin Induce Apoptotic Changes in Mammalian Lymphoid Tissues and Intestine. J. Pathol. 1987, 151, 221–229. [Google Scholar] [CrossRef]
- Rabinovich, G.A.; Toscano, M.A. Turning “sweet” on Immunity: Galectin-Glycan Interactions in Immune Tolerance and Inflammation. Nat. Rev. Immunol. 2009, 9, 338–352. [Google Scholar]
- Chavez, V.; Garcia, A.; Ynalvez, R. Plant Lectin-Induced Apoptosis in Cancer Cells: A Mini-Review. Bios 2020, 91, 79–89. [Google Scholar] [CrossRef]
- Parshenkov, A.; Hennet, T. Glycosylation-Dependent Induction of Programmed Cell Death in Murine Adenocarcinoma Cells. Front. Immunol. 2022, 13, 797759. [Google Scholar] [CrossRef]
- Fu, L.L.; Zhou, C.C.; Yao, S.; Yu, J.Y.; Liu, B.; Bao, J.K. Plant Lectins: Targeting Programmed Cell Death Pathways as Antitumor Agents. Int. J. Biochem. Cell Biol. 2011, 43, 1442–1449. [Google Scholar]
- Carvalho, E.V.M.M.; Oliveira, W.F.; Coelho, L.C.B.B.; Correia, M.T.S. Lectins as Mitosis Stimulating Factors: Briefly Reviewed. Life Sci. 2018, 207, 152–157. [Google Scholar]
- Gerlach, J.Q.; Flannery, A.; Kilcoyne, M. Lectins and Their Applications in Biomedical Research. In Translational Glycobiology in Human Health and Disease; Elsevier: Amsterdam, The Netherlands, 2024; pp. 37–53. [Google Scholar]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Whitaker, R.H.; Cook, J.G. Stress Relief Techniques: P38 MAPK Determines the Balance of Cell Cycle and Apoptosis Pathways. Biomolecules 2021, 11, 1444. [Google Scholar] [CrossRef]
- Liu, T.; Wu, L.; Wang, D.; Wang, H.; Chen, J.; Yang, C.; Bao, J.; Wu, C. Role of Reactive Oxygen Species-Mediated MAPK and NF-ΚB Activation in Polygonatum cyrtonema Lectin-Induced Apoptosis and Autophagy in Human Lung Adenocarcinoma A549 Cells. J. Biochem. 2016, 160, 315–324. [Google Scholar] [CrossRef]
- Liu, B.; Cheng, Y.; Bian, H.J.; Bao, J.K. Molecular Mechanisms of Polygonatum cyrtonema Lectin-Induced Apoptosis and Autophagy in Cancer Cells. Autophagy 2009, 5, 253–255. [Google Scholar] [CrossRef]
- Behera, B.; Mishra, D.; Roy, B.; Devi, K.S.P.; Narayan, R.; Das, J.; Ghosh, S.K.; Maiti, T.K. Abrus precatorius Agglutinin-Derived Peptides Induce ROS-Dependent Mitochondrial Apoptosis through JNK and Akt/P38/P53 Pathways in HeLa Cells. Chem. Biol. Interact. 2014, 222, 97–105. [Google Scholar] [CrossRef]
- Zhuang, Y.; Li, Y.; Li, X.; Xie, Q.; Wu, M. Atg7 Knockdown Augments Concanavalin A-Induced Acute Hepatitis through an ROS-Mediated P38/MAPK Pathway. PLoS ONE 2016, 11, e0149754. [Google Scholar] [CrossRef]
- Yau, T.; Dan, X.; Ng, C.; Ng, T. Lectins with Potential for Anti-Cancer Therapy. Molecules 2015, 20, 3791–3810. [Google Scholar] [CrossRef]
- Mazalovska, M.; Kouokam, J.C. Plant-Derived Lectins as Potential Cancer Therapeutics and Diagnostic Tools. Biomed. Res. Int. 2020, 2020, 1–13. [Google Scholar] [CrossRef]
- Konozy, E.H.E.; Osman, M.E.F.M. Plant Lectin: A Promising Future Anti-Tumor Drug. Biochimie 2022, 202, 136–145. [Google Scholar] [CrossRef]
- Da Silva, T.; Oliveira-Brito, P.; Gonçalves, T.; Vendruscolo, P.; Roque-Barreira, M. ArtinM Mediates Murine T Cell Activation and Induces Cell Death in Jurkat Human Leukemic T Cells. Int. J. Mol. Sci. 2017, 18, 1400. [Google Scholar] [CrossRef]
- Dupuis, G.; Bastin, B. Lectin Interactions with the Jurkat Leukemic T-Cell Line: Quantitative Binding Studies and Interleukin-2 Production. J. Leukoc. Biol. 1988, 43, 238–247. [Google Scholar] [CrossRef]
- Perišić Nanut, M.; Žurga, S.; Konjar, Š.; Prunk, M.; Kos, J.; Sabotič, J. The Fungal Clitocybe nebularis Lectin Binds Distinct Cell Surface Glycoprotein Receptors to Induce Cell Death Selectively in Jurkat Cells. FASEB J. 2022, 36, e22215. [Google Scholar] [CrossRef]
- Steff, A.-M.; Fortin, M.; Philippoussis, F.; Lesage, S.; Arguin, C.; Johnson, P.; Hugo, P. A Cell Death Pathway Induced by Antibody-Mediated Cross-Linking of CD45 on Lymphocytes. Crit. Rev. Immunol. 2003, 23, 421–440. [Google Scholar] [CrossRef]
- Benoist, H.; Culerrier, R.; Poiroux, G.; Ségui, B.; Jauneau, A.; Van Damme, E.J.M.; Peumans, W.J.; Barre, A.; Rougé, P. Two Structurally Identical Mannose-Specific Jacalin-Related Lectins Display Different Effects on Human T Lymphocyte Activation and Cell Death. J. Leukoc. Biol. 2009, 86, 103–114. [Google Scholar] [CrossRef]
- Poiroux, G.; Barre, A.; Simplicien, M.; Pelofy, S.; Segui, B.; Van Damme, E.J.M.; Rougé, P.; Benoist, H. Morniga-G, a T/Tn-Specific Lectin, Induces Leukemic Cell Death via Caspase and DR5 Receptor-Dependent Pathways. Int. J. Mol. Sci. 2019, 20, 230. [Google Scholar] [CrossRef]
- Pujari, R.; Eligar, S.M.; Kumar, N.; Barkeer, S.; Reddy, V.; Swamy, B.M.; Inamdar, S.R.; Shastry, P. Rhizoctonia bataticola Lectin (RBL) Induces Caspase-8-Mediated Apoptosis in Human T-Cell Leukemia Cell Lines but Not in Normal CD3 and CD34 Positive Cells. PLoS ONE 2013, 8, e79311. [Google Scholar] [CrossRef]
- Sundblad, V.; Morosi, L.G.; Geffner, J.R.; Rabinovich, G.A. Galectin-1: A Jack-of-All-Trades in the Resolution of Acute and Chronic Inflammation. J. Immunol. 2017, 199, 3721–3730. [Google Scholar] [CrossRef]
- Perillo, N.L.; Pace, K.E.; Seilhamer, J.J.; Baum, L.G. Apoptosis of T Cells Mediated by Galectin-1. Nature 1995, 378, 736–739. [Google Scholar] [CrossRef]
- Guo, Y.; Shen, R.; Yu, L.; Zheng, X.; Cui, R.; Song, Y.; Wang, D. Roles of Galectin-3 in the Tumor Microenvironment and Tumor Metabolism (Review). Oncol. Rep. 2020, 44, 1799–1809. [Google Scholar] [CrossRef]
- Nakahara, S.; Oka, N.; Raz, A. On the Role of Galectin-3 in Cancer Apoptosis. Apoptosis 2005, 10, 267–275. [Google Scholar] [CrossRef]
- Pugliese, G.; Iacobini, C.; Pesce, C.M.; Menini, S. Galectin-3: An Emerging All-out Player in Metabolic Disorders and Their Complications. Glycobiology 2015, 25, 136–150. [Google Scholar] [CrossRef]
- Mayerhofer, P.U.; Cook, J.P.; Wahlman, J.; Pinheiro, T.T.J.; Moore, K.A.H.; Lord, J.M.; Johnson, A.E.; Roberts, L.M. Ricin A Chain Insertion into Endoplasmic Reticulum Membranes Is Triggered by a Temperature Increase to 37 °C. J. Biol. Chem. 2009, 284, 10232–10242. [Google Scholar] [CrossRef]
- Carruthers, V.B.; Giddings, O.K.; Sibley, L.D. Secretion of Micronemal Proteins Is Associated with Toxoplasma Invasion of Host Cells. Cell Microbiol. 1999, 1, 225–235. [Google Scholar] [CrossRef]
- Blumenschein, T.M.A.; Friedrich, N.; Childs, R.A.; Saouros, S.; Carpenter, E.P.; Campanero-Rhodes, M.A.; Simpson, P.; Chai, W.; Koutroukides, T.; Blackman, M.J.; et al. Atomic Resolution Insight into Host Cell Recognition by Toxoplasma gondii. EMBO J. 2007, 26, 2808–2820. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Souza, I.E.L.; Roque-Barreira, M.-C.; Panunto-Castelo, A. Recombinant Microneme Proteins MIC1 and MIC4 from Toxoplasma gondii Cause Cytotoxic Effects in the Human Jurkat T-Lymphocyte Cell Line. Pathogens 2025, 14, 372. https://doi.org/10.3390/pathogens14040372
Souza IEL, Roque-Barreira M-C, Panunto-Castelo A. Recombinant Microneme Proteins MIC1 and MIC4 from Toxoplasma gondii Cause Cytotoxic Effects in the Human Jurkat T-Lymphocyte Cell Line. Pathogens. 2025; 14(4):372. https://doi.org/10.3390/pathogens14040372
Chicago/Turabian StyleSouza, Igor E. L., Maria-Cristina Roque-Barreira, and Ademilson Panunto-Castelo. 2025. "Recombinant Microneme Proteins MIC1 and MIC4 from Toxoplasma gondii Cause Cytotoxic Effects in the Human Jurkat T-Lymphocyte Cell Line" Pathogens 14, no. 4: 372. https://doi.org/10.3390/pathogens14040372
APA StyleSouza, I. E. L., Roque-Barreira, M.-C., & Panunto-Castelo, A. (2025). Recombinant Microneme Proteins MIC1 and MIC4 from Toxoplasma gondii Cause Cytotoxic Effects in the Human Jurkat T-Lymphocyte Cell Line. Pathogens, 14(4), 372. https://doi.org/10.3390/pathogens14040372