Hepatitis E Virus Shows More Genomic Alterations in Cell Culture than In Vivo

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
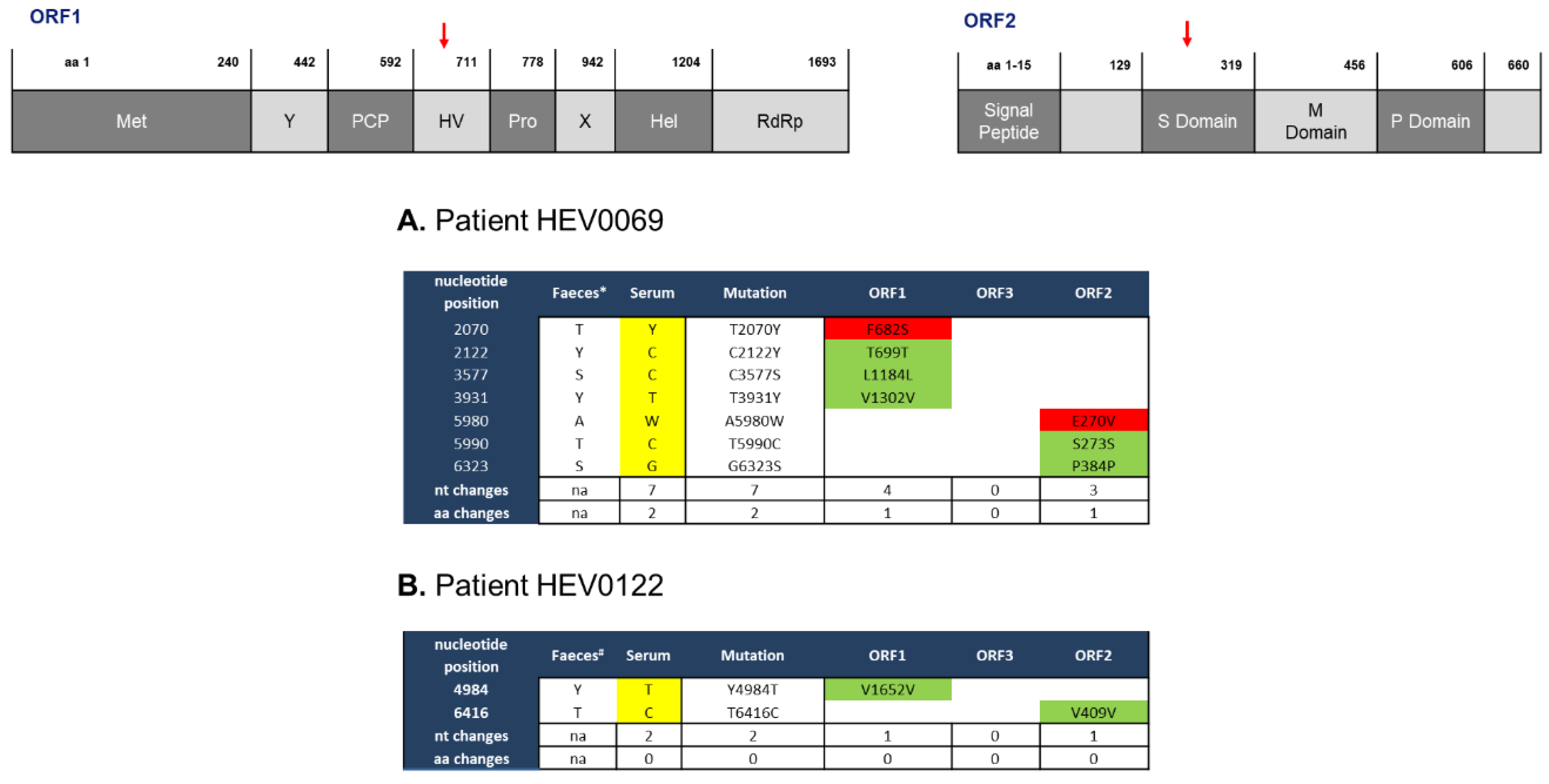
2.1. Feces- and Serum-Derived HEV Sequences Show Mostly Synonymous Mutations
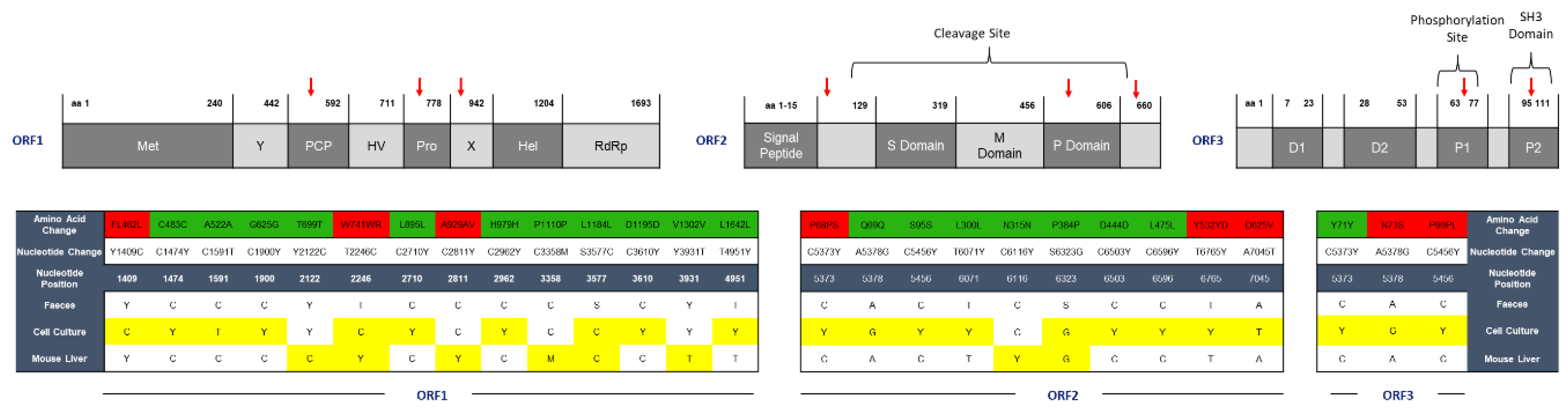
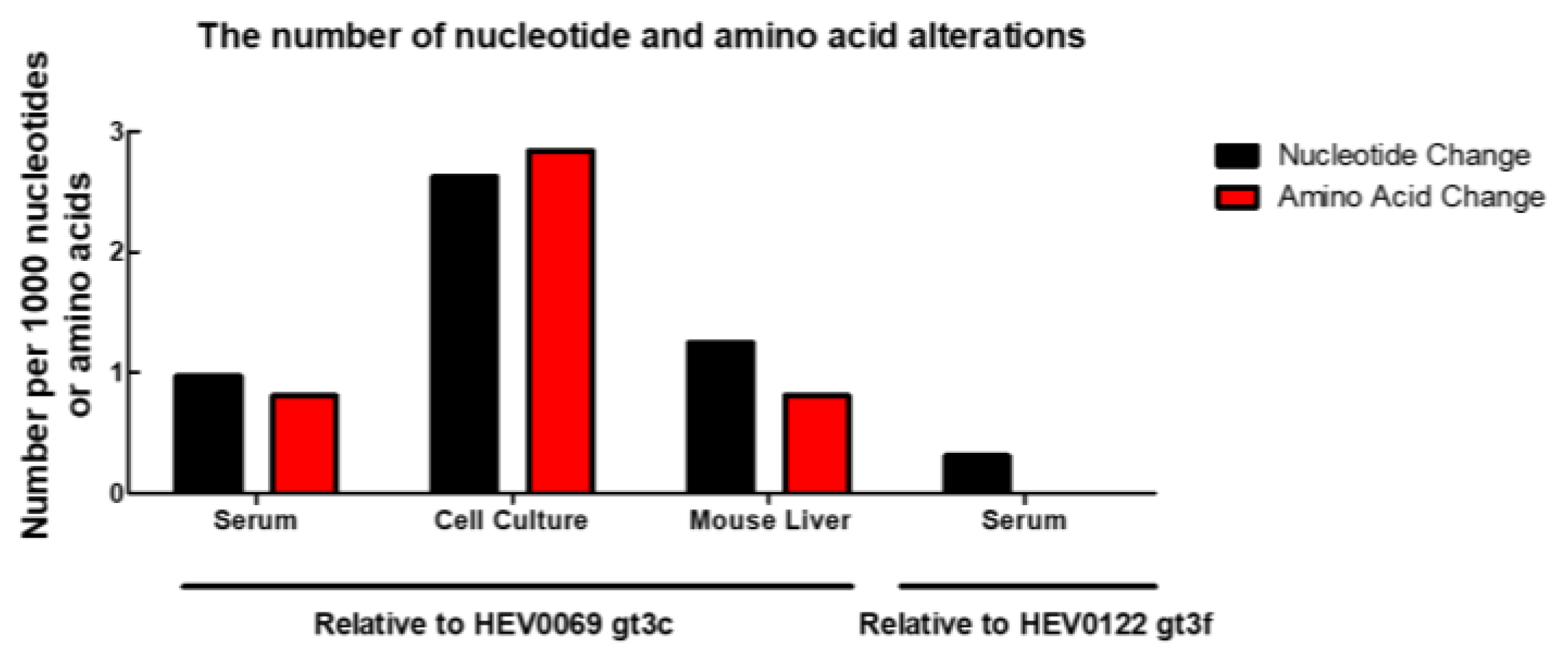
2.2. HEV Adapts to Cell Culture with 7 Nonsynonymous Nucleotide Changes during In Vivo Passage
3. Discussion
4. Materials and Methods
4.1. Virus Isolates
4.2. Cell Culture
4.3. Mouse Origin and In Vivo Experiments
4.4. Viral RNA isolation and cDNA Synthesis
4.5. PCR Amplification
4.6. Fragment Detection and Purification
4.7. Sanger Sequencing
4.8. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Smith, D.B.; Simmonds, P.; Jameel, S.; Emerson, S.U.; Harrison, T.J.; Meng, X.J.; Okamoto, H.; Van der Poel, W.H.; Purdy, M.A. Consensus proposals for classification of the family Hepeviridae. J. Gen. Virol. 2014, 95 Pt 10, 2223–2232. [Google Scholar] [CrossRef]
- Aggarwal, R.; Jameel, S. Hepatitis E. Hepatology 2011, 54, 2218–2226. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Heller, B.; Capuccino, J.M.; Song, B.; Nimgaonkar, I.; Hrebikova, G.; Contreras, J.E.; Ploss, A. Hepatitis E virus ORF3 is a functional ion channel required for release of infectious particles. Proc. Natl. Acad. Sci. USA 2017, 114, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Sitki-Green, D.; Covington, M.; Raab-Traub, N. Compartmentalization and transmission of multiple epstein-barr virus strains in asymptomatic carriers. J. Virol. 2003, 77, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, S.; Perez-Del-Pulgar, S.; Carrion, J.A.; Costa, J.; Gonzalez, P.; Massaguer, A.; Fondevila, C.; Garcia-Valdecasas, J.C.; Navasa, M.; Forns, X. Hepatitis C virus compartmentalization and infection recurrence after liver transplantation. Am. J. Transplant. 2009, 9, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Philpott, S.; Burger, H.; Tsoukas, C.; Foley, B.; Anastos, K.; Kitchen, C.; Weiser, B. Human immunodeficiency virus type 1 genomic RNA sequences in the female genital tract and blood: Compartmentalization and intrapatient recombination. J. Virol. 2005, 79, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Ignacio, C.C.; Torriani, F.; Havlir, D.; Fitch, N.J.; Richman, D.D. In vivo compartmentalization of human immunodeficiency virus: Evidence from the examination of pol sequences from autopsy tissues. J. Virol. 1997, 71, 2059–2071. [Google Scholar]
- Rozera, G.; Abbate, I.; Vlassi, C.; Giombini, E.; Lionetti, R.; Selleri, M.; Zaccaro, P.; Bartolini, B.; Corpolongo, A.; D’Offizi, G.; et al. Quasispecies tropism and compartmentalization in gut and peripheral blood during early and chronic phases of HIV-1 infection: Possible correlation with immune activation markers. Clin. Microbiol. Infect. 2014, 20, O157–O166. [Google Scholar] [CrossRef]
- Harouaka, D.; Engle, R.E.; Wollenberg, K.; Diaz, G.; Tice, A.B.; Zamboni, F.; Govindarajan, S.; Alter, H.; Kleiner, D.E.; Farci, P. Diminished viral replication and compartmentalization of hepatitis C virus in hepatocellular carcinoma tissue. Proc. Natl. Acad. Sci. USA 2016, 113, 1375–1380. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL clinical practice guidelines on hepatitis E virus infection. J. Hepatol. 2018, 68, 1256–1271. [Google Scholar] [CrossRef]
- Abravanel, F.; Lhomme, S.; Rostaing, L.; Kamar, N.; Izopet, J. Protracted fecal shedding of HEV during ribavirin therapy predicts treatment relapse. Clin. Infect. Dis. 2015, 60, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Lhomme, S.; Kamar, N.; Nicot, F.; Ducos, J.; Bismuth, M.; Garrigue, V.; Petitjean-Lecherbonnier, J.; Ollivier, I.; Alessandri-Gradt, E.; Goria, O.; et al. Mutation in the hepatitis E virus polymerase and outcome of Ribavirin therapy. Antimicrob. Agents Chemother. 2015, 60, 1608–1614. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Takahashi, M.; Kusano, E.; Okamoto, H. Development and evaluation of an efficient cell-culture system for Hepatitis E virus. J. Gen. Virol. 2007, 88 Pt 3, 903–911. [Google Scholar] [CrossRef]
- Emerson, S.U.; Nguyen, H.; Graff, J.; Stephany, D.A.; Brockington, A.; Purcell, R.H. In vitro replication of hepatitis E virus (HEV) genomes and of an HEV replicon expressing green fluorescent protein. J. Virol. 2004, 78, 4838–4846. [Google Scholar] [CrossRef] [PubMed]
- Van de Garde, M.D.; Pas, S.D.; van der Net, G.; de Man, R.A.; Osterhaus, A.D.; Haagmans, B.L.; Boonstra, A.; Vanwolleghem, T. Hepatitis E Virus (HEV) genotype 3 infection of human liver chimeric mice as a model for chronic HEV infection. J. Virol. 2016, 90, 4394–4401. [Google Scholar] [CrossRef]
- Sayed, I.M.; Verhoye, L.; Cocquerel, L.; Abravanel, F.; Foquet, L.; Montpellier, C.; Debing, Y.; Farhoudi, A.; Wychowski, C.; Dubuisson, J.; et al. Study of hepatitis E virus infection of genotype 1 and 3 in mice with humanised liver. Gut 2017, 66, 920–929. [Google Scholar] [CrossRef]
- Allweiss, L.; Gass, S.; Giersch, K.; Groth, A.; Kah, J.; Volz, T.; Rapp, G.; Schöbel, A.; Lohse, A.W.; Polywka, S.; et al. Human liver chimeric mice as a new model of chronic hepatitis E virus infection and preclinical drug evaluation. J. Hepatol. 2016, 64, 1033–1040. [Google Scholar] [CrossRef]
- Van de Garde, M.D.B.; Pas, S.D.; van Oord, G.W.; Gama, L.; Choi, Y.; de Man, R.A.; Boonstra, A.; Vanwolleghem, T. Interferon-alpha treatment rapidly clears Hepatitis E virus infection in humanized mice. Sci. Rep. 2017, 7, 8267. [Google Scholar] [CrossRef]
- Smith, D.B.; Simmonds, P.; Izopet, J.; Oliveira-Filho, E.F.; Ulrich, R.G.; Johne, R.; Koenig, M.; Jameel, S.; Harrison, T.J.; Meng, X.J.; et al. Proposed reference sequences for hepatitis E virus subtypes. J. Gen. Virol. 2016, 97, 537–542. [Google Scholar] [CrossRef]
- Grandadam, M.; Tebbal, S.; Caron, M.; Siriwardana, M.; Larouze, B.; Koeck, J.L.; Buisson, Y.; Enouf, V.; Nicand, E. Evidence for hepatitis E virus quasispecies. J. Gen. Virol. 2004, 85 Pt 11, 3189–3194. [Google Scholar] [CrossRef]
- Aprea, G.; Amoroso, M.G.; Di Bartolo, I.; D’Alessio, N.; Di Sabatino, D.; Boni, A.; Cioffi, B.; D’Angelantonio, D.; Scattolini, S.; De Sabato, L. Molecular detection and phylogenetic analysis of hepatitis E virus strains circulating in wild boars in south-central Italy. Transbound. Emerg. Dis. 2018, 65, e25–e31. [Google Scholar] [CrossRef] [PubMed]
- Billam, P.; Sun, Z.F.; Meng, X.J. Analysis of the complete genomic sequence of an apparently avirulent strain of avian hepatitis E virus (avian HEV) identified major genetic differences compared with the prototype pathogenic strain of avian HEV. J. Gen. Virol. 2007, 88 Pt 5, 1538–1544. [Google Scholar] [CrossRef]
- Córdoba, L.; Huang, Y.W.; Opriessnig, T.; Harral, K.K.; Beach, N.M.; Finkielstein, C.V.; Emerson, S.U.; Meng, X.J. Three amino acid mutations (F51L, T59A, and S390L) in the capsid protein of the hepatitis E virus collectively contribute to virus attenuation. J. Virol. 2011, 85, 5338–5349. [Google Scholar] [CrossRef] [PubMed]
- Debing, Y.; Gisa, A.; Dallmeier, K.; Pischke, S.; Bremer, B.; Manns, M.; Wedemeyer, H.; Suneetha, P.V.; Neyts, J. A mutation in the hepatitis E virus RNA polymerase promotes its replication and associates with ribavirin treatment failure in organ transplant recipients. Gastroenterology 2014, 147, 1008–1011.e7. [Google Scholar] [CrossRef] [PubMed]
- Gouttenoire, J.; Szkolnicka, D.; Moradpour, D. Treatment of chronic hepatitis E with ribavirin: Lessons from deep sequencing. Gut 2016, 65, 1583–1584. [Google Scholar] [CrossRef]
- Lu, L.; Li, C.; Hagedorn, C.H. Phylogenetic analysis of global hepatitis E virus sequences: Genetic diversity, subtypes and zoonosis. Rev. Med. Virol. 2006, 16, 5–36. [Google Scholar] [CrossRef]
- Todt, D.; Gisa, A.; Radonic, A.; Nitsche, A.; Behrendt, P.; Suneetha, P.V.; Pischke, S.; Bremer, B.; Brown, R.J.; Manns, M.P. In vivo evidence for ribavirin-induced mutagenesis of the hepatitis E virus genome. Gut 2016, 65, 1733–1743. [Google Scholar] [CrossRef]
- Risso-Ballester, J.; Cuevas, J.M.; Sanjuan, R. Genome-wide estimation of the spontaneous mutation rate of human adenovirus 5 by high.-fidelity deep sequencing. PLoS Pathog. 2016, 12, e1006013. [Google Scholar] [CrossRef]
- Bagdassarian, E.; Doceul, V.; Pellerin, M.; Demange, A.; Meyer, L.; Jouvenet, N.; Pavio, N. The Amino-Terminal region. of hepatitis E virus ORF1 containing a Methyltransferase (Met.) and a Papain-Like Cysteine Protease (PCP) domain counteracts type I interferon response. Viruses 2018, 10, 726. [Google Scholar] [CrossRef]
- Koning, L.; Pas, S.D.; de Man, R.A.; Balk, A.H.; de Knegt, R.J.; ten Kate, F.J.; Osterhaus, A.D.; van der Eijk, A.A. Clinical implications of chronic hepatitis E virus infection in heart transplant recipients. J. Heart Lung Transplant. 2013, 32, 78–85. [Google Scholar] [CrossRef]
- Vanwolleghem, T.; Libbrecht, L.; Hansen, B.E.; Desombere, I.; Roskams, T.; Meuleman, P.; Leroux-Roels, G. Factors determining successful engraftment of hepatocytes and susceptibility to hepatitis B and C virus infection in uPA-SCID mice. J. Hepatol. 2010, 53, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Pas, S.D.; de Man, R.A.; Mulders, C.; Balk, A.H.; van Hal, P.T.; Weimar, W.; Koopmans, M.P.; Osterhaus, A.D.; van der Eijk, A.A. Hepatitis E virus infection among solid organ transplant recipients, The Netherlands. Emerg. Infect. Dis. 2012, 18, 869–872. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Chimeno, M.; Forero, J.E.; Echevarría, J.M.; Muñoz-Bellido, J.L.; Vázquez-López, L.; Morago, L.; García-Galera, M.C.; Avellón, A. Full coding hepatitis E virus genotype 3 genome amplification method. J. Virol. Methods 2016, 230, 18–23. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sari, G.; van de Garde, M.D.B.; van Schoonhoven, A.; Voermans, J.J.C.; van der Eijk, A.A.; de Man, R.A.; Boonstra, A.; Vanwolleghem, T.; Pas, S.D. Hepatitis E Virus Shows More Genomic Alterations in Cell Culture than In Vivo. Pathogens 2019, 8, 255. https://doi.org/10.3390/pathogens8040255
Sari G, van de Garde MDB, van Schoonhoven A, Voermans JJC, van der Eijk AA, de Man RA, Boonstra A, Vanwolleghem T, Pas SD. Hepatitis E Virus Shows More Genomic Alterations in Cell Culture than In Vivo. Pathogens. 2019; 8(4):255. https://doi.org/10.3390/pathogens8040255
Chicago/Turabian StyleSari, Gulce, Martijn D.B. van de Garde, Anne van Schoonhoven, Jolanda J.C. Voermans, Annemiek A. van der Eijk, Robert A. de Man, Andre Boonstra, Thomas Vanwolleghem, and Suzan D. Pas. 2019. "Hepatitis E Virus Shows More Genomic Alterations in Cell Culture than In Vivo" Pathogens 8, no. 4: 255. https://doi.org/10.3390/pathogens8040255
APA StyleSari, G., van de Garde, M. D. B., van Schoonhoven, A., Voermans, J. J. C., van der Eijk, A. A., de Man, R. A., Boonstra, A., Vanwolleghem, T., & Pas, S. D. (2019). Hepatitis E Virus Shows More Genomic Alterations in Cell Culture than In Vivo. Pathogens, 8(4), 255. https://doi.org/10.3390/pathogens8040255