Whole Genome Characterization and Evolutionary Analysis of G1P[8] Rotavirus A Strains during the Pre- and Post-Vaccine Periods in Mozambique (2012–2017)

 , ,
, ,  , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Genome Constellation
2.2. Phylogenetic Analyses
2.2.1. Sequence Analyses of VP7 and VP4
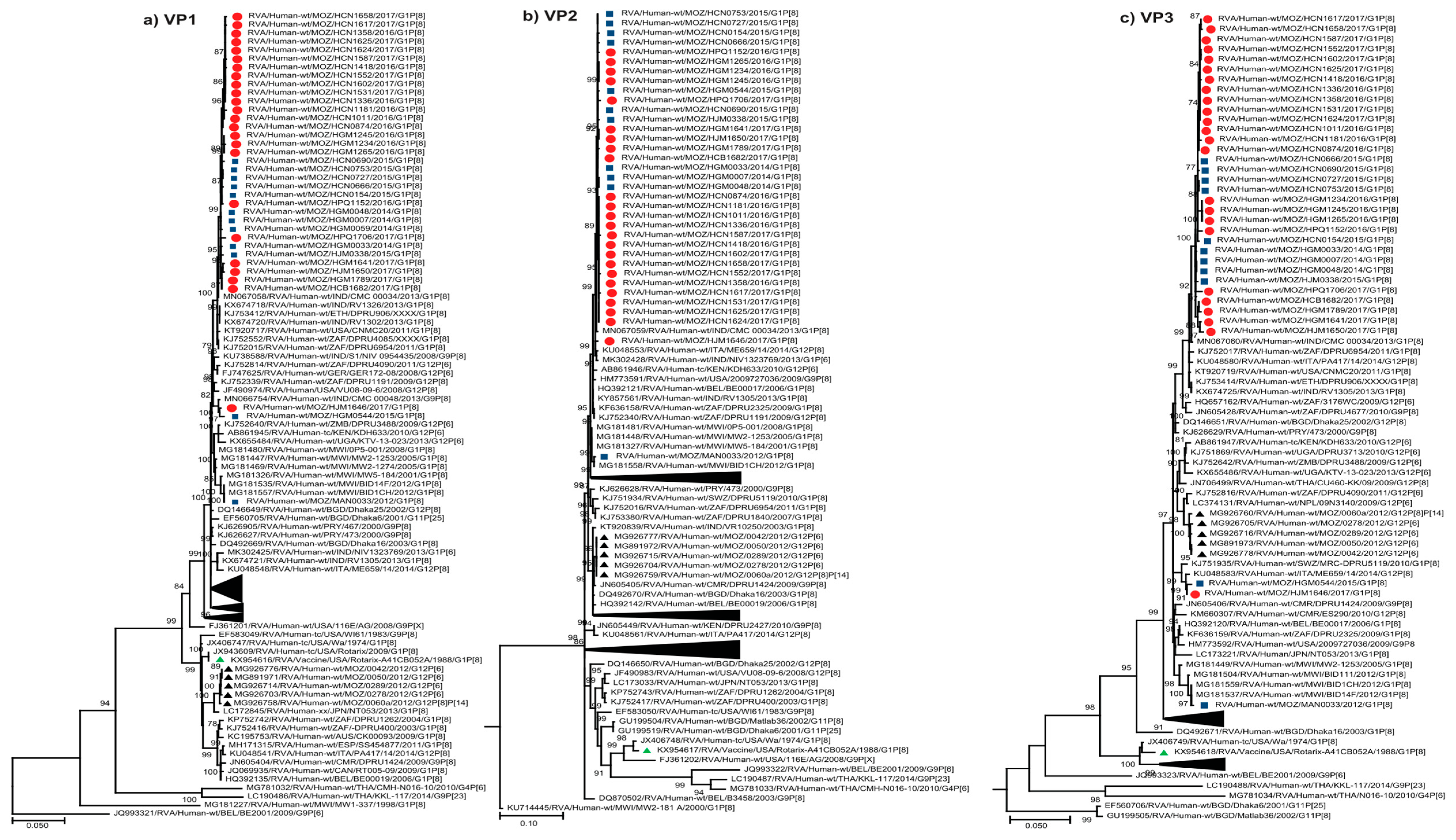
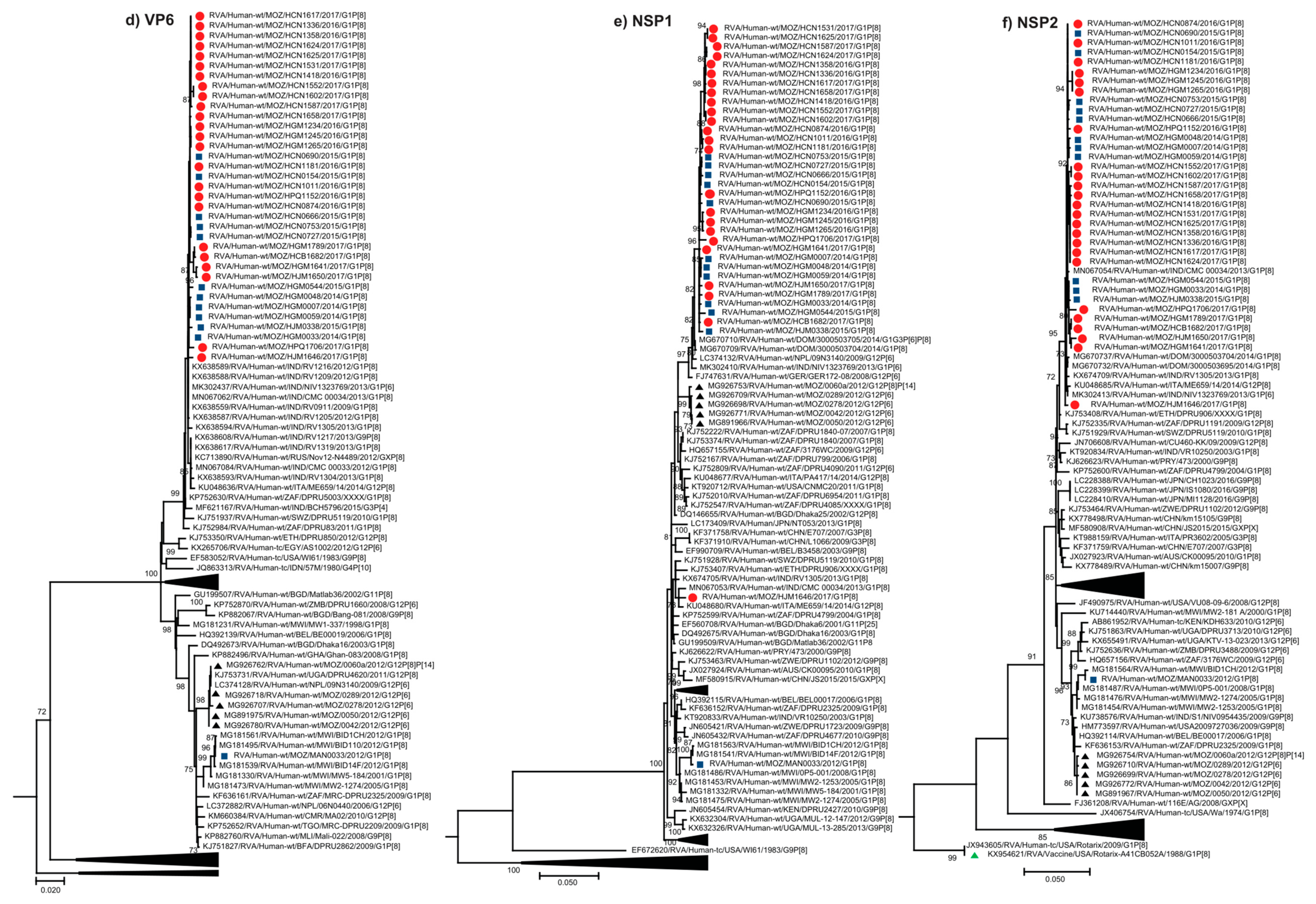
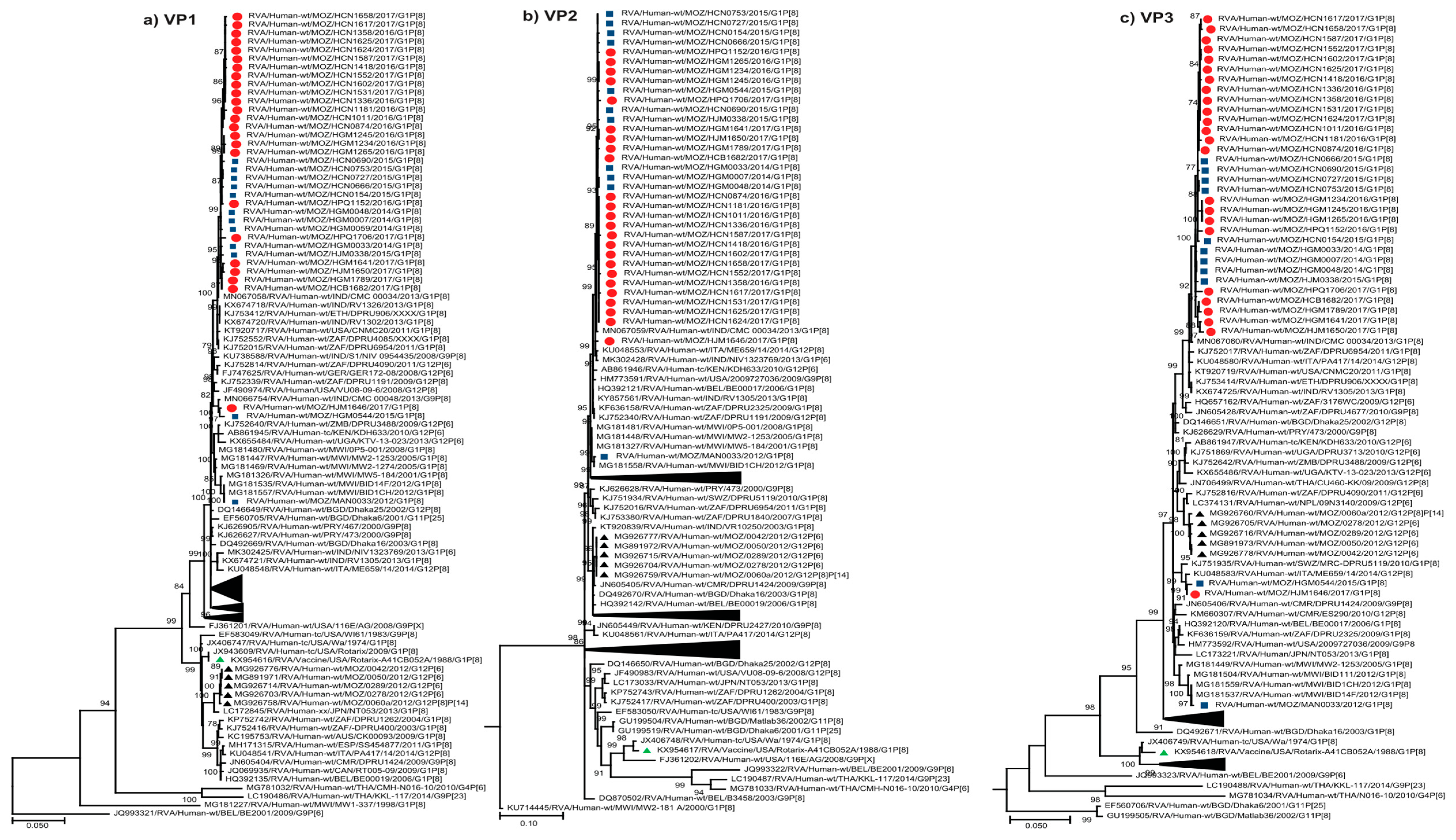
2.2.2. Sequence Analyses of VP1-VP3 and VP6
2.2.3. Sequence Analyses of NSP1-NSP5/6
2.3. Evolutionary Analysis of VP7 and VP4 Genes
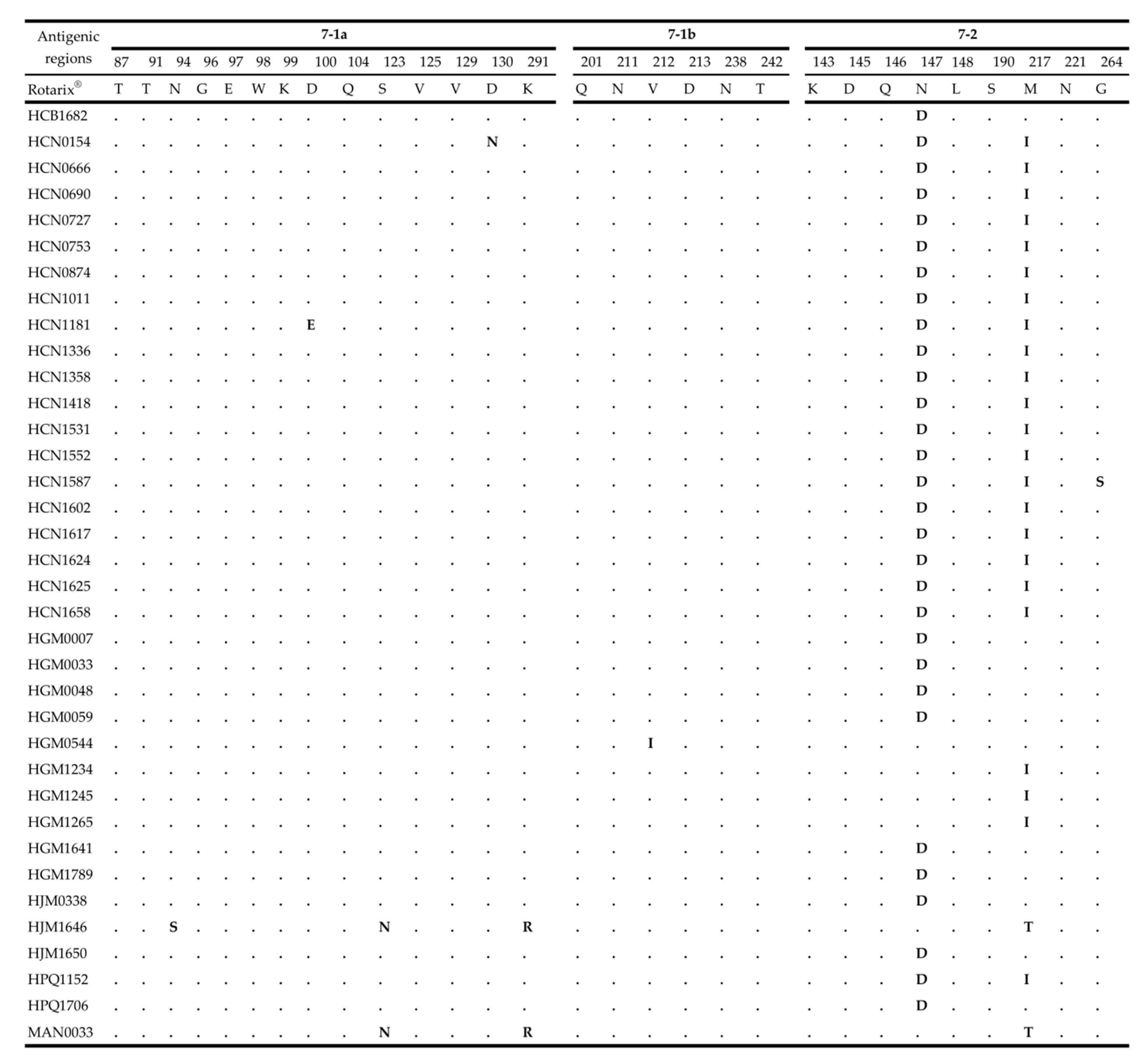
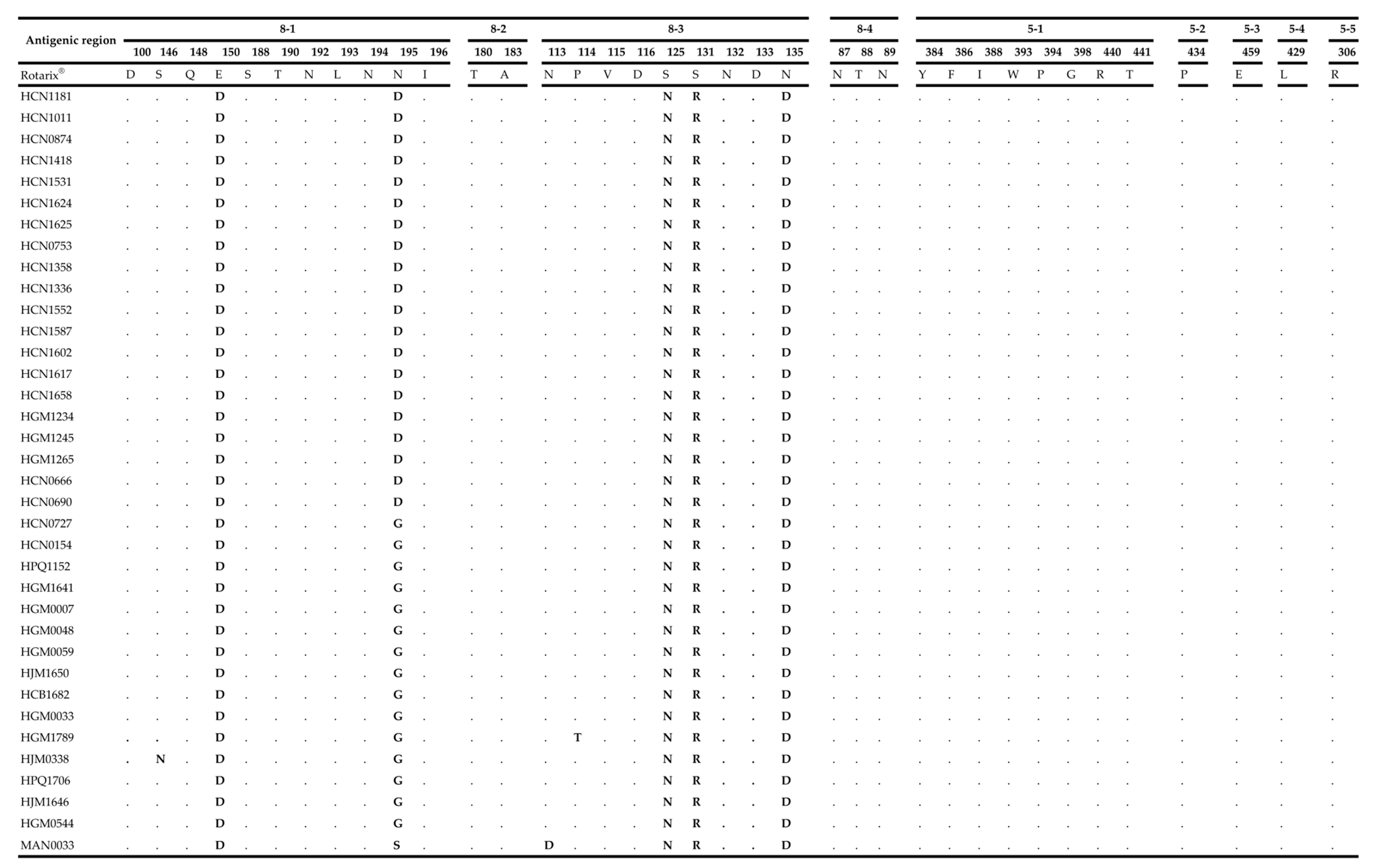
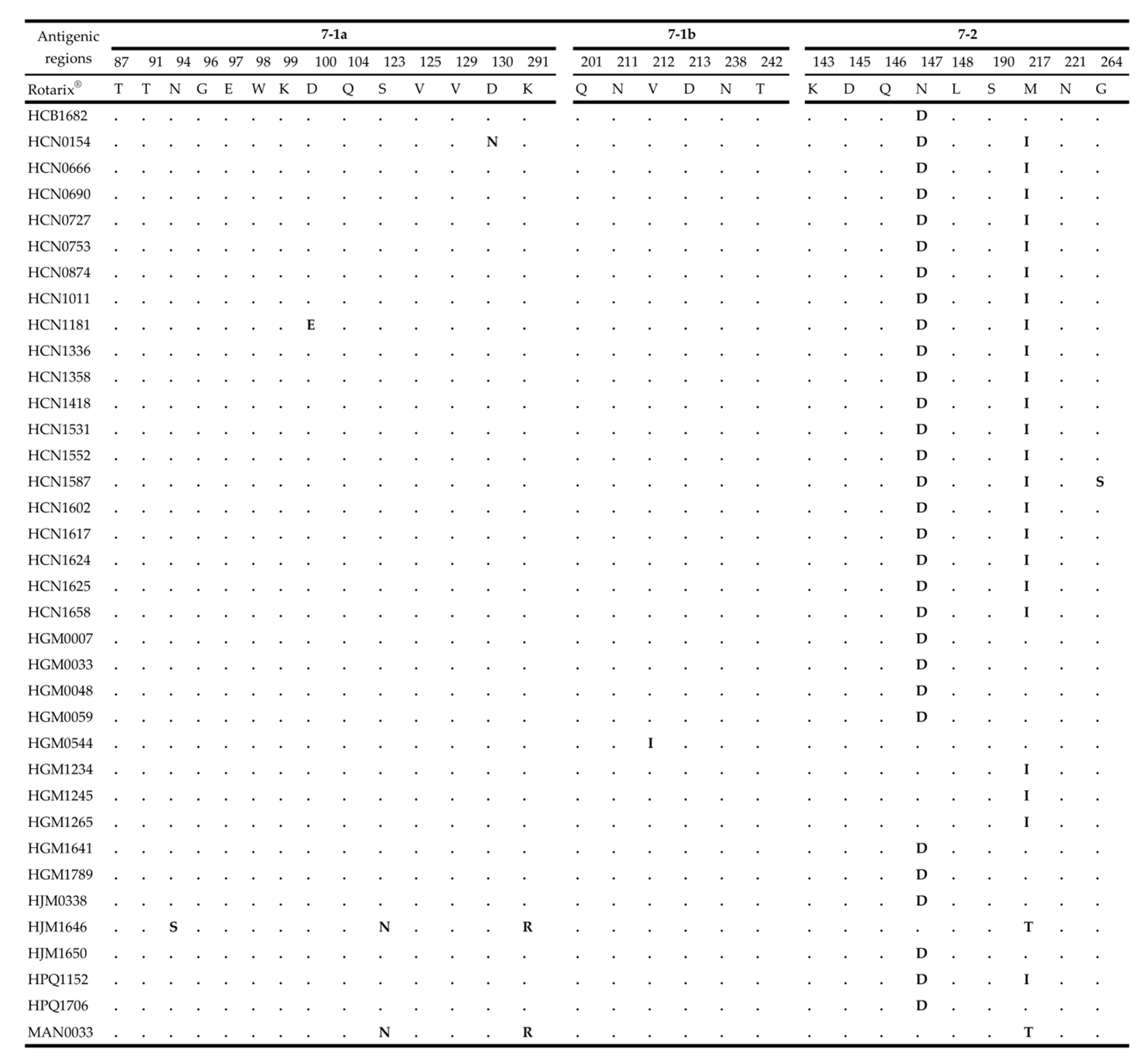
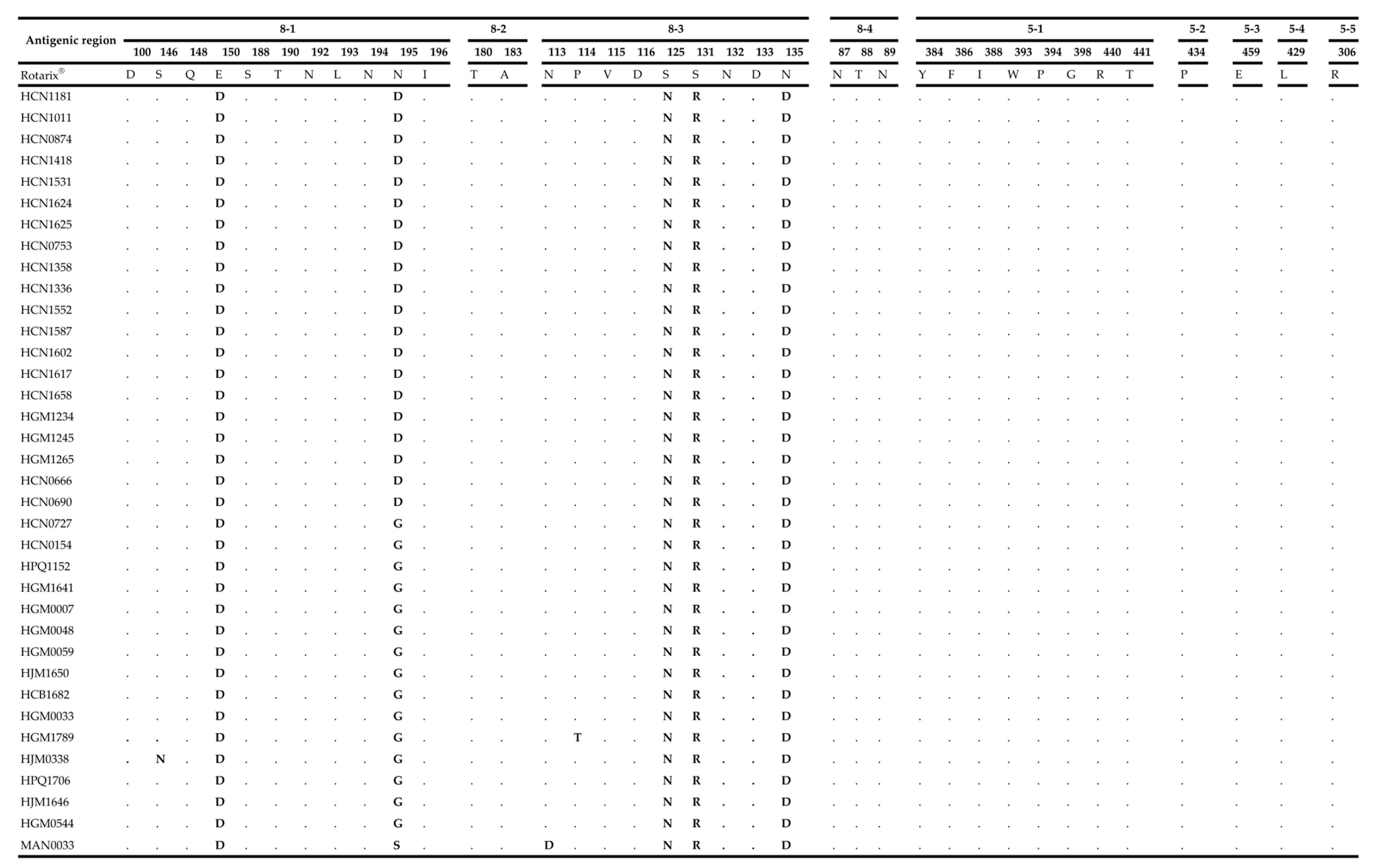
2.4. Comparative Analysis of Neutralizing Antigenic Epitopes of the VP7 and VP4 Genes of Mozambican Strains and the Rotarix® Vaccine Strain
3. Discussion
4. Materials and Methods
4.1. Ethics Approval
4.2. Sample Collection
4.3. RNA Extraction and cDNA Synthesis
4.4. Next Generation Sequencing
4.5. Data Analyses
4.6. Phylogenetic Analysis
4.7. Evolutionary Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tate, J.E.; Burton, A.H.; Boschi-Pinto, C.; Parashar, U.D. Global, Regional, and National Estimates of Rotavirus Mortality in Children <5 Years of Age, 2000–2013. Clin. Infect. Dis. 2016, 62 (Suppl. 2), S96–S105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troeger, C.; Khalil, I.A.; Rao, P.C.; Cao, S.; Blacker, B.F.; Ahmed, T.; Armah, G.; Bines, J.E.; Brewer, T.G.; Colombara, D.V.; et al. Rotavirus Vaccination and the Global Burden of Rotavirus Diarrhea Among Children Younger Than 5 Years. JAMA Pediatr. 2018, 172, 958–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estes, M.K.; Cohen, J. Rotavirus gene structure and function. Microbiol. Rev. 1989, 53, 410–449. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, H.; Estes, M.; Prasad, B.V. Emerging themes in rotavirus cell entry, genome organization, transcription and replication. Virus Res. 2004, 101, 67–81. [Google Scholar] [CrossRef]
- Desselberger, U. Rotaviruses. Virus Res. 2014, 190, 75–96. [Google Scholar] [CrossRef] [Green Version]
- De Deus, N.; João, E.; Cuamba, A.; Cassocera, M.; Luís, L.; Acácio, S.; Mandomando, I.; Augusto, O.; Page, N. Epidemiology of Rotavirus Infection in Children from a Rural and Urban Area, in Maputo, Southern Mozambique, before Vaccine Introduction. J. Trop. Pediatr. 2017, 64, 141–145. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Ciarlet, M.; McDonald, S.M.; Attoui, H.; Banyai, K.; Brister, J.R.; Buesa, J.; Esona, M.D.; Estes, M.K.; Gentsch, J.R.; et al. Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG). Arch. Virol. 2011, 156, 1397–1413. [Google Scholar] [CrossRef] [Green Version]
- Rotavirus Classification Working Group. Virus Classification. Available online: https://rega.kuleuven.be/cev/viralmetagenomics/virus-classification (accessed on 5 June 2020).
- Matthijnssens, J.; Ciarlet, M.; Heiman, E.; Arijs, I.; Delbeke, T.; McDonald, S.M.; Palombo, E.A.; Iturriza-Gómara, M.; Maes, P.; Patton, J.T.; et al. Full Genome-Based Classification of Rotaviruses Reveals a Common Origin between Human Wa-Like and Porcine Rotavirus Strains and Human DS-1-Like and Bovine Rotavirus Strains. J. Virol. 2008, 82, 3204–3219. [Google Scholar] [CrossRef] [Green Version]
- Mwenda, J.M.; Tate, J.E.; Parashar, U.D.; Mihigo, R.; Agócs, M.; Serhan, F.; Nshimirimana, D. African Rotavirus Surveillance Network. Pediatr. Infect. Dis. J. 2014, 33, S6–S8. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Van Ranst, M. Genotype constellation and evolution of group A rotaviruses infecting humans. Curr. Opin. Virol. 2012, 2, 426–433. [Google Scholar] [CrossRef]
- World Health Organization. WHO Prequalifies New Rotavirus Vaccine. Available online: http://www.who.int/medicines/news/2018/prequalified_new-rotavirus_vaccine/en/ (accessed on 5 June 2020).
- Burke, R.M.; Tate, J.E.; Kirkwood, C.D.; Steele, A.D.; Parashar, U.D. Current and new rotavirus vaccines. Curr. Opin. Infect. Dis. 2019, 32, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Langa, J.S.; Thompson, R.; Arnaldo, P.; Resque, H.R.; Rose, T.; Enosse, S.M.; Fialho, A.; De Assis, R.M.S.; Da Silva, M.F.M.; Leite, J.P.G. Epidemiology of rotavirus A diarrhea in Chókwè, Southern Mozambique, from February to September, 2011. J. Med. Virol. 2016, 88, 1751–1758. [Google Scholar] [CrossRef] [PubMed]
- João, E.D.; Strydom, A.; O’Neill, H.G.; Cuamba, A.; Cassocera, M.; Acácio, S.; Mandomando, I.; Motanyane, L.; Page, N.; De Deus, N. Rotavirus A strains obtained from children with acute gastroenteritis in Mozambique, 2012–2013: G and P genotypes and phylogenetic analysis of VP7 and partial VP4 genes. Arch. Virol. 2017, 163, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Deus, N.; Chilaúle, J.J.; Cassocera, M.; Bambo, M.; Langa, J.S.; Sitoe, E.; Chissaque, A.; Anapakala, E.; Sambo, J.; Guimarães, E.L.; et al. Early impact of rotavirus vaccination in children less than five years of age in Mozambique. Vaccine 2018, 36, 7205–7209. [Google Scholar] [CrossRef]
- World Health Organization. WHO and UNICEF Estimates of National Immunization Coverage. Available online: http://www.who.int/immunization/monitoring_surveillance/routine/coverage/en/index4.html (accessed on 1 April 2020).
- João, E.; Munlela, B.; Chissaque, A.; Chilaúle, J.; Langa, J.S.; Augusto, O.; Boene, S.; Anapakala, E.; Sambo, J.; Guimarães, E.; et al. Molecular Epidemiology of Rotavirus A Strains Pre- and Post-Vaccine (Rotarix®) Introduction in Mozambique, 2012–2019: Emergence of Genotypes G3P[4] and G3P[8]. Pathogens 2020, 9, 671. [Google Scholar] [CrossRef]
- Strydom, A.; Motanyane, L.; Nyaga, M.M.; João, E.D.; Cuamba, A.; Mandomando, I.; Cassocera, M.; De Deus, N.; O’Neill, H.G. Whole-genome characterization of G12 rotavirus strains detected in Mozambique reveals a co-infection with a GXP[14] strain of possible animal origin. J. Gen. Virol. 2019, 100, 932–937. [Google Scholar] [CrossRef]
- Strydom, A.; João, E.D.; Motanyane, L.; Nyaga, M.M.; Potgieter, A.C.; Cuamba, A.; Mandomando, I.; Cassocera, M.; De Deus, N.; O’Neill, H.G. Whole genome analyses of DS-1-like Rotavirus A strains detected in children with acute diarrhoea in southern Mozambique suggest several reassortment events. Infect. Genet. Evol. 2019, 69, 68–75. [Google Scholar] [CrossRef]
- Le, V.P.; Chung, Y.-C.; Kim, K.; Chung, S.-I.; Lim, I.; Kim, W. Genetic variation of prevalent G1P[8] human rotaviruses in South Korea. J. Med. Virol. 2010, 82, 886–896. [Google Scholar] [CrossRef]
- Arista, S.; Giammanco, G.M.; De Grazia, S.; Ramirez, S.; Biundo, C.L.; Colomba, C.; Cascio, A.; Martella, V. Heterogeneity and Temporal Dynamics of Evolution of G1 Human Rotaviruses in a Settled Population. J. Virol. 2006, 80, 10724–10733. [Google Scholar] [CrossRef] [Green Version]
- Zeller, M.; Patton, J.T.; Heylen, E.; De Coster, S.; Ciarlet, M.; Van Ranst, M.; Matthijnssens, J. Genetic Analyses Reveal Differences in the VP7 and VP4 Antigenic Epitopes between Human Rotaviruses Circulating in Belgium and Rotaviruses in Rotarix and RotaTeq. J. Clin. Microbiol. 2012, 50, 966–976. [Google Scholar] [CrossRef] [Green Version]
- Ianiro, G.; Delogu, R.; Fiore, L.; Ruggeri, F.M. Genetic variability of VP7, VP4, VP6 and NSP4 genes of common human G1P[8] rotavirus strains circulating in Italy between 2010 and 2014. Virus Res. 2016, 220, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Almeida, T.N.V.; De Sousa, T.T.; Da Silva, R.A.; Fiaccadori, F.S.; Souza, M.; Badr, K.R.; de Paula Cardoso, D.d.D. Phylogenetic analysis of G1P[8] and G12P[8] rotavirus A samples obtained in the pre- and post-vaccine periods, and molecular modeling of VP4 and VP7 proteins. Acta Trop. 2017, 173, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Magagula, N.B.; Esona, M.D.; Nyaga, M.M.; Stucker, K.M.; Halpin, R.A.; Stockwell, T.B.; Seheri, M.L.; Steele, A.D.; Wentworth, D.E.; Mphahlele, M.J. Whole genome analyses of G1P[8] rotavirus strains from vaccinated and non-vaccinated South African children presenting with diarrhea. J. Med. Virol. 2015, 87, 79–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damanka, S.; Kwofie, S.; Dennis, F.E.; Lartey, B.L.; Agbemabiese, C.A.; Doan, Y.H.; Adiku, T.K.; Katayama, K.; Enweronu-Laryea, C.C.; Armah, G.E. Whole genome characterization and evolutionary analysis of OP354-like P[8] Rotavirus A strains isolated from Ghanaian children with diarrhoea. PLoS ONE 2019, 14, e0218348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jere, K.C.; Chaguza, C.; Bar-Zeev, N.; Lowe, J.; Peno, C.; Kumwenda, B.; Nakagomi, O.; Tate, J.E.; Parashar, U.D.; Heyderman, R.S.; et al. Emergence of Double- and Triple-Gene Reassortant G1P[8] Rotaviruses Possessing a DS-1-Like Backbone after Rotavirus Vaccine Introduction in Malawi. J. Virol. 2017, 92, e01246-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Aoki, S.T.; Settembre, E.C.; Trask, S.D.; Greenberg, H.B.; Harrison, S.C.; Dormitzer, P.R. Structure of Rotavirus Outer-Layer Protein VP7 Bound with a Neutralizing Fab. Science 2009, 324, 1444–1447. [Google Scholar] [CrossRef] [Green Version]
- Dormitzer, P.R.; Sun, Z.J.; Wagner, G.; Harrison, S.C. The rhesus rotavirus VP4 sialic acid binding domain has a galectin fold with a novel carbohydrate binding site. EMBO J. 2002, 21, 885–897. [Google Scholar] [CrossRef]
- Dormitzer, P.R.; Nason, E.B.; Prasad, B.V.V.; Harrison, S.C. Structural rearrangements in the membrane penetration protein of a non-enveloped virus. Nat. Cell Biol. 2004, 430, 1053–1058. [Google Scholar] [CrossRef]
- Da Silva, M.F.M.; Rose, T.L.; Gómez, M.M.; Carvalho-Costa, F.A.; Fialho, A.M.; De Assis, R.M.; Sde Andrade, J.d.S.R.; Volotão, E.D.M.; Leite, J.P.G. G1P[8] species A rotavirus over 27 years–Pre- and post-vaccination eras–in Brazil: Full genomic constellation analysis and no evidence for selection pressure by Rotarix® vaccine. Infect. Genet. Evol. 2015, 30, 206–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeller, M.; Donato, C.; Trovão, N.S.; Cowley, D.; Heylen, E.; Donker, N.C.; McAllen, J.K.; Akopov, A.; Kirkness, E.F.; Lemey, P.; et al. Genome-Wide Evolutionary Analyses of G1P[8] Strains Isolated Before and After Rotavirus Vaccine Introduction. Genome Biol. Evol. 2015, 7, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Rasebotsa, S.; Mwangi, P.N.; Mogotsi, M.T.; Sabiu, S.; Magagula, N.B.; Rakau, K.; Uwimana, J.; Mutesa, L.; Muganga, N.; Murenzi, D.; et al. Whole genome and in-silico analyses of G1P[8] rotavirus strains from pre- and post-vaccination periods in Rwanda. Sci. Rep. 2020, 10, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Mwangi, P.N.; Mogotsi, M.; Rasebotsa, S.P.; Seheri, M.L.; Mphahlele, M.J.; Ndze, V.N.; Dennis, F.E.; Jere, K.C.; Nyaga, M.M. Uncovering the First Atypical DS-1-like G1P[8] Rotavirus Strains That Circulated during Pre-Rotavirus Vaccine Introduction Era in South Africa. Pathogens 2020, 9, 391. [Google Scholar] [CrossRef]
- Donato, C.M.; Ch’Ng, L.S.; Boniface, K.F.; Crawford, N.W.; Buttery, J.P.; Lyon, M.; Bishop, R.F.; Kirkwood, C.D. Identification of Strains of RotaTeq Rotavirus Vaccine in Infants With Gastroenteritis Following Routine Vaccination. J. Infect. Dis. 2012, 206, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Gower, C.M.; Dunning, J.; Nawaz, S.; Allen, D.; Ramsay, M.E.; Ladhani, S.N. Vaccine-derived rotavirus strains in infants in England. Arch. Dis. Child. 2019, 105, 553–557. [Google Scholar] [CrossRef]
- Arora, R.; Chitambar, S. Full genomic analysis of Indian G1P[8] rotavirus strains. Infect. Genet. Evol. 2011, 11, 504–511. [Google Scholar] [CrossRef]
- Kulkarni, R.; Arora, R.; Arora, R.; Chitambar, S.D. Sequence analysis of VP7 and VP4 genes of G1P[8] rotaviruses circulating among diarrhoeic children in Pune, India: A comparison with Rotarix and RotaTeq vaccine strains. Vaccine 2014, 32, A75–A83. [Google Scholar] [CrossRef] [Green Version]
- Gouvea, V.; Glass, R.I.; Woods, P.; Taniguchi, K.; Clark, H.F.; Forrester, B.; Fang, Z.Y. Polymerase chain reaction amplification and typing of rotavirus nucleic acid from stool specimens. J. Clin. Microbiol. 1990, 28, 276–282. [Google Scholar] [CrossRef] [Green Version]
- Gentsch, J.R.; Glass, R.I.; Woods, P.; Gouvea, V.; Gorziglia, M.; Flores, J.; Das, B.K.; Bhan, M.K. Identification of group A rotavirus gene 4 types by polymerase chain reaction. J. Clin. Microbiol. 1992, 30, 1365–1373. [Google Scholar] [CrossRef] [Green Version]
- Potgieter, A.C.; Page, N.A.; Liebenberg, J.; Wright, I.M.; Landt, O.; Van Dijk, A. Improved strategies for sequence-independent amplification and sequencing of viral double-stranded RNA genomes. J. Gen. Virol. 2009, 90, 1423–1432. [Google Scholar] [CrossRef] [PubMed]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef] [PubMed]
- Maes, P.; Matthijnssens, J.; Rahman, M.; Van Ranst, M. RotaC: A web-based tool for the complete genome classification of group A rotaviruses. BMC Microbiol. 2009, 9, 238. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Hazkani-Covo, E.; Graur, D. A Comparative Analysis of numt Evolution in Human and Chimpanzee. Mol. Biol. Evol. 2006, 24, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Hasegawa, M.; Kishino, H.; Yano, T.-A. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munlela, B.; João, E.D.; Donato, C.M.; Strydom, A.; Boene, S.S.; Chissaque, A.; Bauhofer, A.F.L.; Langa, J.; Cassocera, M.; Cossa-Moiane, I.; et al. Whole Genome Characterization and Evolutionary Analysis of G1P[8] Rotavirus A Strains during the Pre- and Post-Vaccine Periods in Mozambique (2012–2017). Pathogens 2020, 9, 1026. https://doi.org/10.3390/pathogens9121026
Munlela B, João ED, Donato CM, Strydom A, Boene SS, Chissaque A, Bauhofer AFL, Langa J, Cassocera M, Cossa-Moiane I, et al. Whole Genome Characterization and Evolutionary Analysis of G1P[8] Rotavirus A Strains during the Pre- and Post-Vaccine Periods in Mozambique (2012–2017). Pathogens. 2020; 9(12):1026. https://doi.org/10.3390/pathogens9121026
Chicago/Turabian StyleMunlela, Benilde, Eva D. João, Celeste M. Donato, Amy Strydom, Simone S. Boene, Assucênio Chissaque, Adilson F. L. Bauhofer, Jerónimo Langa, Marta Cassocera, Idalécia Cossa-Moiane, and et al. 2020. "Whole Genome Characterization and Evolutionary Analysis of G1P[8] Rotavirus A Strains during the Pre- and Post-Vaccine Periods in Mozambique (2012–2017)" Pathogens 9, no. 12: 1026. https://doi.org/10.3390/pathogens9121026
APA StyleMunlela, B., João, E. D., Donato, C. M., Strydom, A., Boene, S. S., Chissaque, A., Bauhofer, A. F. L., Langa, J., Cassocera, M., Cossa-Moiane, I., Chilaúle, J. J., O’Neill, H. G., & de Deus, N. (2020). Whole Genome Characterization and Evolutionary Analysis of G1P[8] Rotavirus A Strains during the Pre- and Post-Vaccine Periods in Mozambique (2012–2017). Pathogens, 9(12), 1026. https://doi.org/10.3390/pathogens9121026