Detection of Helicobacter pylori Microevolution and Multiple Infection from Gastric Biopsies by Housekeeping Gene Amplicon Sequencing

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Samples and Amplicon Sequences
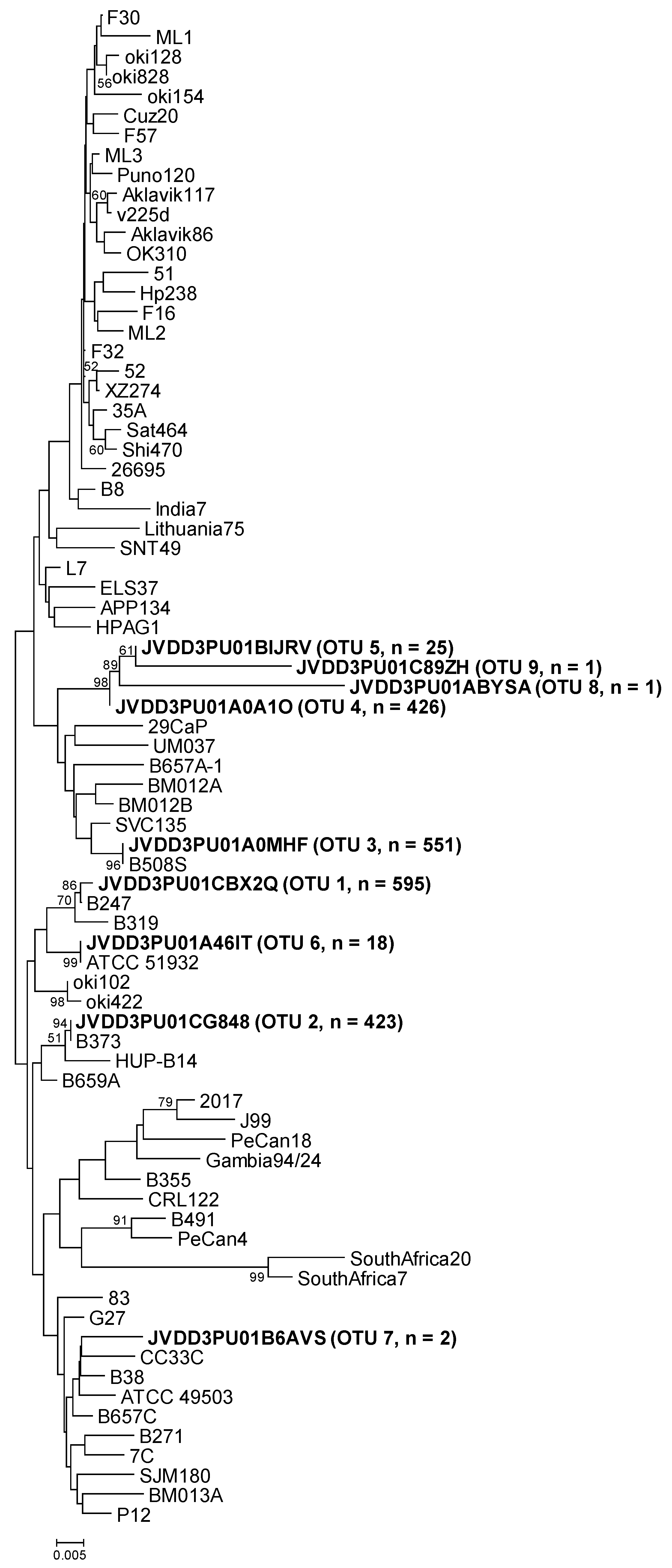
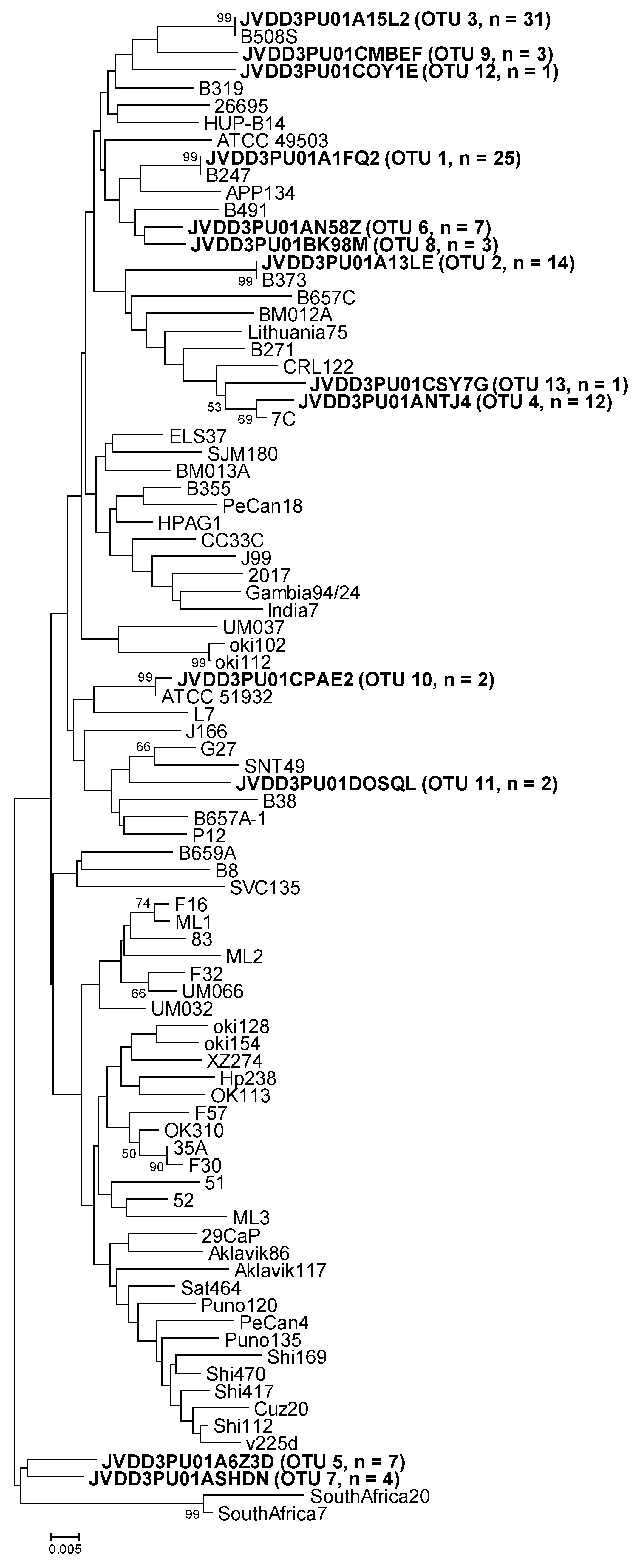
2.2. Operational Taxonomic Units (OTUs) Assignment and Distribution
2.3. Amplicon Sequence Identification
3. Discussion
4. Materials and Methods
4.1. Gastric Biopsies
4.2. DNA Extraction and PCR Amplification
4.3. Preparation of Libraries
4.4. Data Processing
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moodley, Y.; Linz, B.; Bond, R.P.; Nieuwoudt, M.; Soodyall, H.; Schlebusch, C.M.; Bernhöft, S.; Hale, J.; Suerbaum, S.; Mugisha, L.; et al. Age of the association between Helicobacter pylori and man. PLoS Pathog. 2012, 8, e1002693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, R.; Shiota, S.; Yamaoka, Y. Molecular epidemiology, population genetics, and pathogenic role of Helicobacter pylori. Infect. Genet. Evol. 2012, 12, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Piqué, N.; Palau, M.; Berlanga, M.; Miñana-Galbis, D. Advances in the research of new genetic markers for the detection of Helicobacter pylori infection. In Recent Advances in Pharmaceutical Sciences VI; Transworld Research Network: Kerala, India, 2016; pp. 165–188. [Google Scholar]
- Hashi, K.; Imai, C.; Yahara, K.; Tahmina, K.; Hayashi, T.; Azuma, T.; Miyabe-Nishiwaki, T.; Sato, H.; Matsuoka, M.; Niimi, S.; et al. Evaluating the origin and virulence of a Helicobacter pylori cagA-positive strain isolated from a non-human primate. Sci. Rep. 2018, 8, 15981. [Google Scholar] [CrossRef] [PubMed]
- Correa, P.; Houghton, J. Carcinogenesis of Helicobacter pylori. Gastroenterology 2007, 133, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Buzás, G.M. Benign and malignant gastroduodenal diseases associated with Helicobacter pylori: A narrative review and personal remarks in 2018. Minerva Gastroenterol. Dietol. 2018, 64, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Quaglia, N.C.; Dambrosio, A. Helicobacter pylori: A foodborne pathogen? World J. Gastroenterol. 2018, 24, 3472–3487. [Google Scholar] [CrossRef] [PubMed]
- Kabamba, E.T.; Tuan, V.P.; Yamaoka, Y. Genetic populations and virulence factors of Helicobacter pylori. Infect. Genet. Evol. 2018, 60, 109–116. [Google Scholar] [CrossRef]
- Smith, S.; Fowora, M.; Pellicano, R. Infections with Helicobacter pylori and challenges encountered in Africa. World J. Gastroenterol. 2019, 25, 3183–3195. [Google Scholar] [CrossRef]
- Ben Mansour, K.; Fendri, C.; Battikh, H.; Garnier, M.; Zribi, M.; Jlizi, A.; Burucoa, C. Multiple and mixed Helicobacter pylori infections: Comparison of two epidemiological situations in Tunisia and France. Infect. Genet. Evol. 2016, 37, 43–48. [Google Scholar] [CrossRef]
- Palau, M.; Kulmann, M.; Ramírez-Lázaro, M.J.; Lario, S.; Quílez, M.E.; Campo, R.; Piqué, N.; Calvet, X.; Miñana-Galbis, D. Usefulness of housekeeping genes for the diagnosis of Helicobacter pylori infection, strain discrimination and detection of multiple infection. Helicobacter 2016, 21, 481–487. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Toita, N.; Yokota, S.; Fujii, N.; Konno, M. Clonality Analysis of Helicobacter pylori in patients isolated from several biopsy specimens and gastric juice in a Japanese urban population by random amplified polymorphic DNA fingerprinting. Gastroenterol. Res. Pract. 2013, 2013, 721306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheu, S.M.; Sheu, B.S.; Lu, C.C.; Yang, H.B.; Wu, J.J. Mixed infections of Helicobacter pylori: Tissue tropism and histological significance. Clin. Microbiol. Infect. 2009, 15, 253–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.H.; Huang, J.C.; Chiang-Ni, C.; Li, J.-P.; Wu, L.-T.; Wu, H.-S.; Sun, Y.-C.; Lin, M.-L.; Lee, J.-F.; Lin, H.-J. Mixed infections of Helicobacter pylori isolated from patients with gastrointestinal diseases in Taiwan. Gastroenterol. Res. Pract. 2016, 2016, 7521913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kibria, K.M.; Hossain, M.E.; Sultana, J.; Sarker, S.A.; Bardhan, P.K.; Rahman, M.; Nahar, S. The prevalence of mixed Helicobacter pylori infections in symptomatic and asymptomatic subjects in Dhaka, Bangladesh. Helicobacter 2015, 20, 397–404. [Google Scholar] [CrossRef]
- Raymond, J.; Thiberg, J.M.; Chevalier, C.; Kalach, N.; Bergeret, M.; Labigne, A.; Dauga, C. Genetic and transmission analysis of Helicobacter pylori strains within a family. Emerg. Infect. Dis. 2004, 10, 1816–1821. [Google Scholar] [CrossRef]
- Falush, D.; Kraft, C.; Taylor, N.S.; Correa, P.; Fox, J.G.; Achtman, M.; Suerbaum, S. Recombination and mutation during long-term gastric colonization by Helicobacter pylori: Estimates of clock rates, recombination size, and minimal age. Proc. Natl. Acad. Sci. USA 2001, 98, 15056–15061. [Google Scholar] [CrossRef] [Green Version]
- Morelli, G.; Didelot, X.; Kusecek, B.; Schwarz, S.; Bahlawane, C.; Falush, D.; Suerbaum, S.; Achtman, M. Microevolution of Helicobacter pylori during prolonged infection of single hosts and within families. PLoS Genet. 2010, 6, e1001036. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; Didelot, X.; Wu, Z.; Li, Z.; He, L.; Li, Y.; Ni, M.; You, Y.; Lin, X.; Li, Z.; et al. Progressive genomic convergence of two Helicobacter pylori strains during mixed infection of a patient with chronic gastritis. Gut 2015, 64, 554–561. [Google Scholar] [CrossRef] [Green Version]
- Linz, B.; Windsor, H.M.; McGraw, J.J.; Hansen, L.M.; Gajewski, J.P.; Tomsho, L.P.; Hake, C.M.; Solnick, J.V.; Schuster, S.C.; Marshall, B.J. A mutation burst during the acute phase of Helicobacter pylori infection in humans and rhesus macaques. Nat. Commun. 2014, 5, 4165. [Google Scholar] [CrossRef]
- Lario, S.; Ramírez-Lázaro, M.J.; Sanjuan-Herráez, D.; Brunet-Vega, A.; Pericay, C.; Gombau, L.; Junquera, F.; Quintás, G.; Calvet, X. Plasma sample based analysis of gastric cancer progression using targeted metabolomics. Sci. Rep. 2017, 7, 17774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomics datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| cgt OTUs | B247S Sequences | B373A Sequences | B508S Sequences | B508T Sequences | B601A Sequences | cgt Sequences | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | Unique | Total | Unique | Total | Unique | Total | Unique | Total | Unique | Total | Unique | |
| OTU-01 | 5184 | 592 | − | − | − | − | 3 | 3 | − | − | 5187 | 595 |
| OTU-02 | 1 | 1 | 2416 | 418 | − | − | 4 | 4 | − | − | 2421 | 423 |
| OTU-03 | − | − | − | − | 963 | 230 | 1391 | 321 | − | − | 2354 | 551 |
| OTU-04 | − | − | − | − | − | − | − | − | 1808 | 426 | 1808 | 426 |
| OTU-05 | − | − | − | − | − | − | − | − | 30 | 25 | 30 | 25 |
| OTU-06 | − | − | 6 | 4 | 4 | 3 | 16 | 11 | − | − | 26 | 18 |
| OTU-07 | − | − | − | − | 1 | 1 | − | − | 4 | 1 | 5 | 2 |
| OTU-08 | − | − | − | − | − | − | − | − | 1 | 1 | 1 | 1 |
| OTU-09 | − | − | − | − | − | − | − | − | 1 | 1 | 1 | 1 |
| total | 5185 | 593 (11%) | 2422 | 422 (17%) | 968 | 234 (24%) | 1414 | 339 (24%) | 1844 | 454 (25%) | 11,833 | 2042 (17%) |
| luxS OTUs | B247S Sequences | B373A Sequences | B508S Sequences | B508T Sequences | B601A Sequences | luxS Sequences | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | Unique | Total | Unique | Total | Unique | Total | Unique | Total | Unique | Total | Unique | |
| OTU-01 | 98 | 25 | − | − | − | − | − | − | − | − | 98 | 25 |
| OTU-02 | − | − | 70 | 14 | − | − | − | − | − | − | 70 | 14 |
| OTU-03 | − | − | − | − | 52 | 14 | 107 | 15 | 5 | 2 | 164 | 31 |
| OTU-04 | − | − | − | − | − | − | − | − | 33 | 12 | 33 | 12 |
| OTU-05 | − | − | − | − | 2 | 2 | 1 | 1 | 8 | 4 | 11 | 7 |
| OTU-06 | 7 | 6 | − | − | − | − | 1 | 1 | − | − | 8 | 7 |
| OTU-07 | − | − | 2 | 2 | − | − | 1 | 1 | 1 | 1 | 4 | 4 |
| OTU-08 | 6 | 3 | − | − | − | − | − | − | − | − | 6 | 3 |
| OTU-09 | − | − | − | − | 1 | 1 | 2 | 2 | − | − | 3 | 3 |
| OTU-10 | − | − | − | − | − | − | 2 | 2 | − | − | 2 | 2 |
| OTU-11 | − | − | − | − | 1 | 1 | 1 | 1 | − | − | 2 | 2 |
| OTU-12 | − | − | − | − | − | − | − | − | 1 | 1 | 1 | 1 |
| OTU-13 | − | − | − | − | − | − | − | − | 1 | 1 | 1 | 1 |
| total | 111 | 34 (31%) | 72 | 16 (22%) | 56 | 18 (32%) | 115 | 23 (20%) | 49 | 21 (43%) | 403 | 112 (28%) |
| cgt OTUs | Representative Sequence | Mothur | MEGA7 (% Similarity) | BLAST (% Similarity) |
|---|---|---|---|---|
| OTU-01 | JVDD3PU01CBX2Q | B247 | B247 (99.8) | B247 (100) |
| OTU-02 | JVDD3PU01CG848 | B373A | B373A (100) | B373A (100) |
| OTU-03 | JVDD3PU01A0MHF | B508S | B508S (100) | B508S (100) |
| OTU-04 | JVDD3PU01A0A1O | SVC135 | OTU-05 (99.5), SVC135 (98.6) | SVC135 (99) |
| OTU-05 | JVDD3PU01BIJRV | SVC135 | OTU-04 (99.5), SVC135 (98.2) | SVC135 (98) |
| OTU-06 | JVDD3PU01A46IT | ATCC 51932 | ATCC 51932 (100) | ATCC 51932 (100) |
| OTU-07 | JVDD3PU01B6AVS | B508S | B508S, B657A-4, B38 and OTU-03 (98.4) | B508S, B657A-4 and B38 (98) |
| OTU-08 | JVDD3PU01ABYSA | SVC135 | OTU-04 (95.8), SVC135 (94.3) | SVC135 (98) |
| OTU-09 | JVDD3PU01C89ZH | SVC135 | OTU-05 (97.2), OTU-04 (96.7), SVC135 (95.3) | SVC135 (97) |
| luxS OTUs | Representative Sequence | Mothur | MEGA7 (% Similarity) | BLAST (% Similarity) |
|---|---|---|---|---|
| OTU-01 | JVDD3PU01A1FQ2 | B247 | B247 (100) | B247 (100) |
| OTU-02 | JVDD3PU01A13LE | B373A | B373A (100) | B373A (100) |
| OTU-03 | JVDD3PU01A15L2 | B508S | B508S (100) | B508S (100) |
| OTU-04 | JVDD3PU01ANTJ4 | 7C | 7C (99.2) | 7C (99) |
| OTU-05 | JVDD3PU01A6Z3D | oki112 | OTU-07 (97.8), OTU-06, B247 and B319 (96.9) | B247 and B319 (97) |
| OTU-06 | JVDD3PU01AN58Z | B247 | OTU-08 (98.6), B247 and OTU-02 (98.3) | B247 (98) |
| OTU-07 | JVDD3PU01ASHDN | B373A | OTU-05 (97.8), ELS37 (97.2) | ELS37 (97) |
| OTU-08 | JVDD3PU01BK98M | B491 | OTU-06 (98.6), B319 and B491 (97.5) | B319, B491 and ATCC 51932 (97) |
| OTU-09 | JVDD3PU01CMBEF | B508S | B508S and OTU-03 (97.2) | B508S (98) |
| OTU-10 | JVDD3PU01CPAE2 | ATCC 51932 | ATCC 51932 (99.7) | ATCC 51932 (99) |
| OTU-11 | JVDD3PU01DOSQL | G27 | G27 (97.2) | G27 (97) |
| OTU-12 | JVDD3PU01COY1E | 7C | OTU-04 (97.5), 7C, B319, B508S, ELS37, HPAG1, HUP-B14 and OTU-03 (96.6) | 7C, B319, B508S, ELS37, HPAG1 and HUP-B14 (97) |
| OTU-13 | JVDD3PU01CSY7G | B508S | OTU-04 (97.8), 7C (97.5) | 7C (98) |
| Patient ID | Age and Gender | Endoscopic Diagnosis | Anatomical Location of Biopsy Specimen | Histopathological Diagnosis * | Specimen ID |
|---|---|---|---|---|---|
| B247 | 63 y/o male | Gastric cancer | Non-neoplastic stomach tissue | Chronic active gastritis | B247S |
| B373 | 56 y/o male | Duodenal ulcer | Antrum | Intestinal metaplasia | B373A |
| B508 | 77 y/o male | Gastric cancer | Non-neoplastic stomach tissue | Not available | B508S |
| Tumor tissue | Adenocarcinoma | B508T | |||
| B601 | 43 y/o female | Normal | Antrum | Atrophy | B601A |
| Gene | Fusion Primer | Sequence (Adaptor–key–MID–Template-Specific Sequence) |
|---|---|---|
| cgt | cgt-252-B247S | CCATCTCATCCCTGCGTGTCTCCGACTCAGAGACGCACTCGGCTTTTAAGGGAGCGGATA |
| cgt-252-B373A | CCATCTCATCCCTGCGTGTCTCCGACTCAGAGCACTGTAGGGCTTTTAAGGGAGCGGATA | |
| cgt-252-B508S | CCATCTCATCCCTGCGTGTCTCCGACTCAGACGAGTGCGTGGCTTTTAAGGGAGCGGATA | |
| cgt-252-B508T | CCATCTCATCCCTGCGTGTCTCCGACTCAGACGCTCGACAGGCTTTTAAGGGAGCGGATA | |
| cgt-252-B601A | CCATCTCATCCCTGCGTGTCTCCGACTCAGATCAGACACGGGCTTTTAAGGGAGCGGATA | |
| cgt-866 (reverse) | CCTATCCCCTGTGTGCCTTGGCAGTCTCAGATCGCTTCGCTYTCCACATT | |
| luxS | luxS-38-B247S | CCATCTCATCCCTGCGTGTCTCCGACTCAGAGACGCACTCTGGATCACACYAAAGTCAAAG |
| luxS-38-B373A | CCATCTCATCCCTGCGTGTCTCCGACTCAGAGCACTGTAGTGGATCACACYAAAGTCAAAG | |
| luxS-38-B508S | CCATCTCATCCCTGCGTGTCTCCGACTCAGACGAGTGCGTTGGATCACACYAAAGTCAAAG | |
| luxS-38-B508T | CCATCTCATCCCTGCGTGTCTCCGACTCAGACGCTCGACATGGATCACACYAAAGTCAAAG | |
| luxS-38-B601A | CCATCTCATCCCTGCGTGTCTCCGACTCAGATCAGACACGTGGATCACACYAAAGTCAAAG | |
| luxS-466 (reverse) | CCTATCCCCTGTGTGCCTTGGCAGTCTCAGAAACCCCCACTTCAGACCA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palau, M.; Piqué, N.; Comeau, A.M.; Douglas, G.M.; Ramírez-Lázaro, M.J.; Lario, S.; Calvet, X.; Langille, M.G.I.; Miñana-Galbis, D. Detection of Helicobacter pylori Microevolution and Multiple Infection from Gastric Biopsies by Housekeeping Gene Amplicon Sequencing. Pathogens 2020, 9, 97. https://doi.org/10.3390/pathogens9020097
Palau M, Piqué N, Comeau AM, Douglas GM, Ramírez-Lázaro MJ, Lario S, Calvet X, Langille MGI, Miñana-Galbis D. Detection of Helicobacter pylori Microevolution and Multiple Infection from Gastric Biopsies by Housekeeping Gene Amplicon Sequencing. Pathogens. 2020; 9(2):97. https://doi.org/10.3390/pathogens9020097
Chicago/Turabian StylePalau, Montserrat, Núria Piqué, André M. Comeau, Gavin M. Douglas, M. José Ramírez-Lázaro, Sergio Lario, Xavier Calvet, Morgan G. I. Langille, and David Miñana-Galbis. 2020. "Detection of Helicobacter pylori Microevolution and Multiple Infection from Gastric Biopsies by Housekeeping Gene Amplicon Sequencing" Pathogens 9, no. 2: 97. https://doi.org/10.3390/pathogens9020097