
Gut Dysbiosis and Immune System in Atherosclerotic Cardiovascular Disease (ACVD)

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Gut Dysbiosis in ACVD
3. The Immune System and ACVD
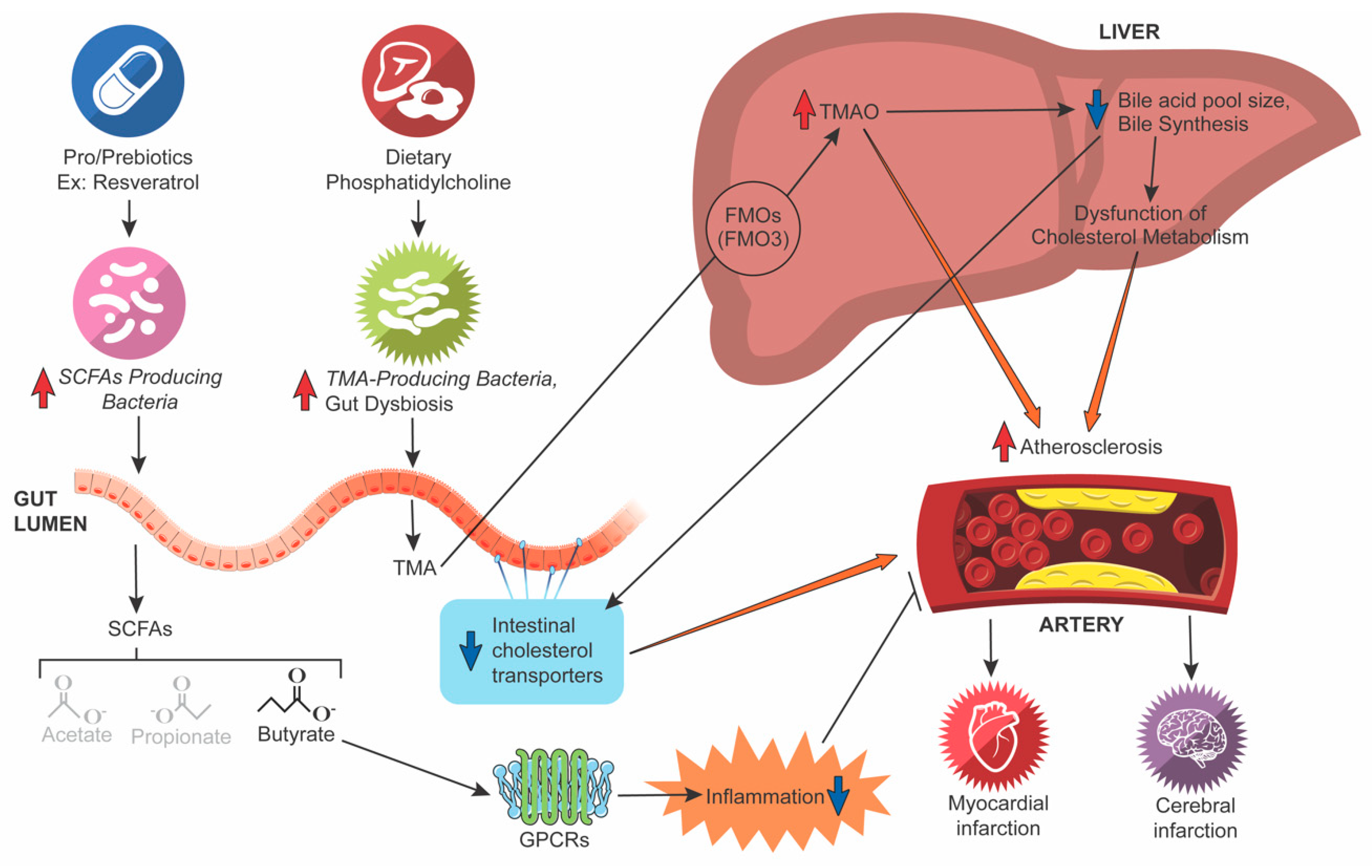
4. The TMA/TMAO Pathway
5. SCFAs and Inflammation
6. Bile Acid (BA) Metabolism
7. Other Factors
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APCs | Antigen-presenting cells |
| ApoE−/− | Apolipoprotein E knockout |
| ACVD | Atherosclerotic cardiovascular disease |
| ANGPTL4 | Angiopoietin-like 4 |
| cAMP | Adenosine 3′,5′-cyclic monophosphate |
| BAs | Bile acids |
| FXR | Bile acid-activated nuclear hormone receptor farnesoid X receptor |
| CVD | Cardiovascular diseases |
| FMOs | Flavin monooxygenases |
| Foxp3 | Forkhead box protein P3 |
| GLP-1 | Glucagon-like peptide 1 |
| GPCRs | G protein-coupled receptors |
| TGR5 | G protein-coupled bile acid receptor |
| HbA1c | Hemoglobin A1c |
| LPS | Lipopolysaccharide |
| LDL | Low-density lipoprotein |
| NF-κB | Nuclear factor-κB |
| OLFR78 | Olfactory receptor 78 |
| PRRs | Pattern recognition receptors |
| PYY | Peptide YY |
| PPARs | Peroxisome proliferator-activated receptors |
| SCFAs | Short-chain fatty acids |
| TMA | Trimethylamine |
| TMAO | Trimethylamine-N-oxide |
| TNF-α | Tumor necrosis factor-alpha |
| Treg | T-regulatory |
| Th17 | T helper 17 cell |
| TLRs | Toll-like receptors |
| T2DM | Type 2 diabetes |
References
- Frostegård, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 117. [Google Scholar] [CrossRef] [Green Version]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Sanz, J.; Fayad, Z.A. Imaging of atherosclerotic cardiovascular disease. Nature 2008, 451, 953–957. [Google Scholar] [CrossRef]
- Arad, Y.; Goodman, K.J.; Roth, M.; Newstein, D.; Guerci, A.D. Coronary calcification, coronary disease risk factors, C-reactive protein, and atherosclerotic cardiovascular disease events: The St. Francis Heart Study. J. Am. Coll. Cardiol. 2005, 46, 158–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, J.W. Whole grains protect against atherosclerotic cardiovascular disease. Proc. Nutr. Soc. 2003, 62, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.Y.; Kim, S.S. Probiotics and Prebiotics: Present Status and Future Perspectives on Metabolic Disorders. Nutrients 2016, 8, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.W.; Kitai, T.; Hazen, S.L. Gut microbiota in cardiovascular health and disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Faith, J.J.; Guruge, J.L.; Charbonneau, M.; Subramanian, S.; Seedorf, H.; Goodman, A.L.; Clemente, J.C.; Knight, R.; Heath, A.C.; Leibel, R.L. The long-term stability of the human gut microbiota. Science 2013, 341. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chávez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y. Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Haraszthy, V.; Zambon, J.; Trevisan, M.; Zeid, M.; Genco, R. Identification of periodontal pathogens in atheromatous plaques. J. Periodontol. 2000, 71, 1554–1560. [Google Scholar] [CrossRef]
- Ziganshina, E.E.; Sharifullina, D.M.; Lozhkin, A.P.; Khayrullin, R.N.; Ignatyev, I.M.; Ziganshin, A.M. Bacterial communities associated with atherosclerotic plaques from Russian individuals with atherosclerosis. PLoS ONE 2016, 11, e0164836. [Google Scholar] [CrossRef] [PubMed]
- Voronina, O.L.; Kunda, M.S.; Ryzhova, N.N.; Aksenova, E.I.; Sharapova, N.E.; Semenov, A.N.; Amelina, E.L.; Chuchalin, A.G.; Gintsburg, A.L. On Burkholderiales order microorganisms and cystic fibrosis in Russia. BMC Genom. 2018, 19, 74. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Arango, L.F.; Barrett, H.L.; Wilkinson, S.A.; Callaway, L.K.; McIntyre, H.D.; Morrison, M.; Dekker Nitert, M. Low dietary fiber intake increases Collinsella abundance in the gut microbiota of overweight and obese pregnant women. Gut Microbes 2018, 9, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, F.H.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wright, K.; Davis, J.M.; Jeraldo, P.; Marietta, E.V.; Murray, J.; Nelson, H.; Matteson, E.L.; Taneja, V. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med. 2016, 8, 436. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Li, Y.; Song, X.; Wang, J.; He, Z.; Zhu, J.; Chen, H.; Yuan, J.; Zhang, X.; Jiang, H.; et al. Both gut microbiota and cytokines act to atherosclerosis in ApoE−/− mice. Microb. Pathog. 2020, 138, 103827. [Google Scholar] [CrossRef]
- Kappel, B.A.; De Angelis, L.; Heiser, M.; Ballanti, M.; Stoehr, R.; Goettsch, C.; Mavilio, M.; Artati, A.; Paoluzi, O.A.; Adamski, J. Cross-omics analysis revealed gut microbiome-related metabolic pathways underlying atherosclerosis development after antibiotics treatment. Mol. Metab. 2020, 36, 100976. [Google Scholar] [CrossRef] [PubMed]
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system. Nature 2011, 474, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakakura, K.; Nakano, M.; Otsuka, F.; Ladich, E.; Kolodgie, F.D.; Virmani, R. Pathophysiology of atherosclerosis plaque progression. Heart Lung Circ. 2013, 22, 399–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut microbiota and immune system interactions. Microorganisms 2020, 8, 1587. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- McLaren, J.E.; Ramji, D.P. Interferon gamma: A master regulator of atherosclerosis. Cytokine Growth Factor Rev. 2009, 20, 125–135. [Google Scholar] [CrossRef]
- Hansson, G.K.; Libby, P. The immune response in atherosclerosis: A double-edged sword. Nat. Rev. Immunol. 2006, 6, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Fadini, G.P.; Simoni, F.; Cappellari, R.; Vitturi, N.; Galasso, S.; de Kreutzenberg, S.V.; Previato, L.; Avogaro, A. Pro-inflammatory monocyte-macrophage polarization imbalance in human hypercholesterolemia and atherosclerosis. Atherosclerosis 2014, 237, 805–808. [Google Scholar] [CrossRef]
- van der Vorst, E.P.; Döring, Y.; Weber, C. Chemokines and their receptors in Atherosclerosis. J. Mol. Med. 2015, 93, 963–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, M.D.; Leaphart, C.; Levy, R.; Prince, J.; Billiar, T.R.; Watkins, S.; Li, J.; Cetin, S.; Ford, H.; Schreiber, A.; et al. Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J. Immunol. 2006, 176, 3070–3079. [Google Scholar] [CrossRef]
- Morikawa, M.; Tsujibe, S.; Kiyoshima-Shibata, J.; Watanabe, Y.; Kato-Nagaoka, N.; Shida, K.; Matsumoto, S. Microbiota of the small intestine is selectively engulfed by phagocytes of the lamina propria and Peyer’s patches. PLoS ONE 2016, 11, e0163607. [Google Scholar] [CrossRef] [Green Version]
- Raetz, C.R.; Reynolds, C.M.; Trent, M.S.; Bishop, R.E. Lipid A modification systems in gram-negative bacteria. Annu. Rev. Biochem. 2007, 76, 295–329. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, M. Structural modifications of bacterial lipopolysaccharide that facilitate gram-negative bacteria evasion of host innate immunity. Front. Immunol. 2013, 4, 109. [Google Scholar] [CrossRef] [Green Version]
- Gu, W.; Wang, Y.; Zeng, L.; Dong, J.; Bi, Q.; Yang, X.; Che, Y.; He, S.; Yu, J. Polysaccharides from Polygonatum kingianum improve glucose and lipid metabolism in rats fed a high fat diet. Biomed. Pharmacother. 2020, 125, 109910. [Google Scholar] [CrossRef] [PubMed]
- Ackah, E.; Yu, J.; Zoellner, S.; Iwakiri, Y.; Skurk, C.; Shibata, R.; Ouchi, N.; Easton, R.M.; Galasso, G.; Birnbaum, M.J.; et al. Akt1/protein kinase Balpha is critical for ischemic and VEGF-mediated angiogenesis. J. Clin. Investig. 2005, 115, 2119–2127. [Google Scholar] [CrossRef]
- Tschopp, O.; Yang, Z.Z.; Brodbeck, D.; Dummler, B.A.; Hemmings-Mieszczak, M.; Watanabe, T.; Michaelis, T.; Frahm, J.; Hemmings, B.A. Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Development 2005, 132, 2943–2954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easton, R.M.; Cho, H.; Roovers, K.; Shineman, D.W.; Mizrahi, M.; Forman, M.S.; Lee, V.M.; Szabolcs, M.; de Jong, R.; Oltersdorf, T.; et al. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol. Cell Biol. 2005, 25, 1869–1878. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Thorvaldsen, J.L.; Chu, Q.; Feng, F.; Birnbaum, M.J. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J. Biol. Chem. 2001, 276, 38349–38352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szelag, M.; Piaszyk-Borychowska, A.; Plens-Galaska, M.; Wesoly, J.; Bluyssen, H.A.R. Targeted inhibition of STATs and IRFs as a potential treatment strategy in cardiovascular disease. Oncotarget 2016, 7, 48788–48812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reustle, A.; Torzewski, M. Role of p38 MAPK in Atherosclerosis and Aortic Valve Sclerosis. Int. J. Mol. Sci. 2018, 19, 3761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muslin, A.J. MAPK signalling in cardiovascular health and disease: Molecular mechanisms and therapeutic targets. Clin. Sci. 2008, 115, 203–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.-D.; Zhu, R.-X.; Bian, Z.-Z.; Pan, X.-T. Improvement of Gut Microbiota by Inhibition of P38 Mitogen-Activated Protein Kinase (MAPK) Signaling Pathway in Rats with Severe Acute Pancreatitis. Med. Sci. Monit. 2019, 25, 4609–4616. [Google Scholar] [CrossRef]
- Craciun, S.; Balskus, E.P. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc. Natl. Acad. Sci. USA 2012, 109, 21307–21312. [Google Scholar] [CrossRef] [Green Version]
- Bartolomaeus, H.; Balogh, A.; Yakoub, M.; Homann, S.; Markó, L.; Höges, S.; Tsvetkov, D.; Krannich, A.; Wundersitz, S.; Avery, E.G.; et al. Short-Chain Fatty Acid Propionate Protects From Hypertensive Cardiovascular Damage. Circulation 2019, 139, 1407–1421. [Google Scholar] [CrossRef]
- Quareshy, M.; Shanmugam, M.; Townsend, E.; Jameson, E.; Bugg, T.D.; Cameron, A.D.; Chen, Y. Structural basis of carnitine monooxygenase CntA substrate specificity, inhibition, and intersubunit electron transfer. J. Biol. Chem. 2021, 296, 100038. [Google Scholar] [CrossRef]
- Zhu, Y.; Jameson, E.; Crosatti, M.; Schäfer, H.; Rajakumar, K.; Bugg, T.D.; Chen, Y. Carnitine metabolism to trimethylamine by an unusual Rieske-type oxygenase from human microbiota. Proc. Natl. Acad. Sci. USA 2014, 111, 4268–4273. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.W.; Wang, Z.; Fan, Y.; Levison, B.; Hazen, J.E.; Donahue, L.M.; Wu, Y.; Hazen, S.L. Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-N-oxide in patients with heart failure: Refining the gut hypothesis. J. Am. Coll. Cardiol. 2014, 64, 1908–1914. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.W.; Hazen, S.L. The contributory role of gut microbiota in cardiovascular disease. J. Clin. Investig. 2014, 124, 4204–4211. [Google Scholar] [CrossRef]
- Hoyles, L.; Fernandez-Real, J.-M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 2018, 24, 1070–1080. [Google Scholar] [CrossRef]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeth, R.A.; Levison, B.S.; Culley, M.K.; Buffa, J.A.; Wang, Z.; Gregory, J.C.; Org, E.; Wu, Y.; Li, L.; Smith, J.D. γ-Butyrobetaine is a proatherogenic intermediate in gut microbial metabolism of L-carnitine to TMAO. Cell Metab. 2014, 20, 799–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Liu, X.; Xu, J.; Xue, C.; Xue, Y.; Wang, Y. Dietary trimethylamine N-oxide exacerbates impaired glucose tolerance in mice fed a high fat diet. J. Biosci. Bioeng. 2014, 118, 476–481. [Google Scholar] [CrossRef]
- Chen, M.; Yi, L.; Zhang, Y.; Zhou, X.; Ran, L.; Yang, J.; Zhu, J.; Zhang, Q.; Mi, M. Resveratrol attenuates trimethylamine-N-oxide (TMAO)-induced atherosclerosis by regulating TMAO synthesis and bile acid metabolism via remodeling of the gut microbiota. MBio 2016, 7, e02210-15. [Google Scholar] [CrossRef] [Green Version]
- Venegas, D.P.; Marjorie, K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.; Faber, K.N.; Hermoso, M.A. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [Green Version]
- Macfarlane, G.; Gibson, G. Microbiological aspects of the production of short-chain fatty acids in the large bowel. Physiol. Clin. Asp. Short-Chain. Fat. Acids 1995, 87–105. Available online: https://centaur.reading.ac.uk/35495/ (accessed on 22 December 2021).
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef] [Green Version]
- Chambers, E.S.; Preston, T.; Frost, G.; Morrison, D.J. Role of gut microbiota-generated short-chain fatty acids in metabolic and cardiovascular health. Curr. Nutr. Rep. 2018, 7, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Zhou, T.; Wang, X.-A.; Tong, X.; Ding, J. Histone deacetylases and atherosclerosis. Atherosclerosis 2015, 240, 355–366. [Google Scholar] [CrossRef]
- Licciardi, P.V.; Ververis, K.; Karagiannis, T.C. Histone deacetylase inhibition and dietary short-chain Fatty acids. ISRN Allergy 2011, 2011, 869647. [Google Scholar] [CrossRef] [Green Version]
- Usami, M.; Kishimoto, K.; Ohata, A.; Miyoshi, M.; Aoyama, M.; Fueda, Y.; Kotani, J. Butyrate and trichostatin A attenuate nuclear factor kappaB activation and tumor necrosis factor alpha secretion and increase prostaglandin E2 secretion in human peripheral blood mononuclear cells. Nutr. Res. 2008, 28, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Trautwein, E.A.; Rieckhoff, D.r.; Erbersdobler, H.F. Dietary inulin lowers plasma cholesterol and triacylglycerol and alters biliary bile acid profile in hamsters. J. Nutr. 1998, 128, 1937–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gäbel, G.; Aschenbach, J.; Müller, F. Transfer of energy substrates across the ruminal epithelium: Implications and limitations. Anim. Health Res. Rev. 2002, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Sukonina, V.; Lookene, A.; Olivecrona, T.; Olivecrona, G. Angiopoietin-like protein 4 converts lipoprotein lipase to inactive monomers and modulates lipase activity in adipose tissue. Proc. Natl. Acad. Sci. USA 2006, 103, 17450–17455. [Google Scholar] [CrossRef] [Green Version]
- Kersten, S.; Mandard, S.; Tan, N.S.; Escher, P.; Metzger, D.; Chambon, P.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Characterization of the fasting-induced adipose factor FIAF, a novel peroxisome proliferator-activated receptor target gene. J. Biol. Chem. 2000, 275, 28488–28493. [Google Scholar] [CrossRef] [Green Version]
- Ferré, P. The biology of peroxisome proliferator-activated receptors: Relationship with lipid metabolism and insulin sensitivity. Diabetes 2004, 53, S43–S50. [Google Scholar] [CrossRef] [Green Version]
- Mattijssen, F.; Alex, S.; Swarts, H.J.; Groen, A.K.; van Schothorst, E.M.; Kersten, S. Angptl4 serves as an endogenous inhibitor of intestinal lipid digestion. Mol. Metab. 2014, 3, 135–144. [Google Scholar] [CrossRef]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, E.; Leonel, A.; Teixeira, L.; Silva, A.; Silva, J.; Pelaez, J.; Capettini, L.; Lemos, V.; Santos, R.; Alvarez-Leite, J. Butyrate impairs atherogenesis by reducing plaque inflammation and vulnerability and decreasing NFκB activation. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 606–613. [Google Scholar] [CrossRef]
- Marques, F.Z.; Nelson, E.; Chu, P.Y.; Horlock, D.; Fiedler, A.; Ziemann, M.; Tan, J.K.; Kuruppu, S.; Rajapakse, N.W.; El-Osta, A.; et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation 2017, 135, 964–977. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [PubMed] [Green Version]
- Pellicciari, R.; Sato, H.; Gioiello, A.; Costantino, G.; Macchiarulo, A.; Sadeghpour, B.M.; Giorgi, G.; Schoonjans, K.; Auwerx, J. Nongenomic actions of bile acids. Synthesis and preliminary characterization of 23-and 6, 23-alkyl-substituted bile acid derivatives as selective modulators for the G-protein coupled receptor TGR5. J. Med. Chem. 2007, 50, 4265–4268. [Google Scholar] [CrossRef]
- Wang, X.X.; Luo, Y.; Wang, D.; Adorini, L.; Pruzanski, M.; Dobrinskikh, E.; Levi, M. A dual agonist of farnesoid X receptor (FXR) and the G protein–coupled receptor TGR5, INT-767, reverses age-related kidney disease in mice. J. Biol. Chem. 2017, 292, 12018–12024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Houten, S.M.; Mataki, C.; Christoffolete, M.A.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 2006, 439, 484–489. [Google Scholar] [CrossRef]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki-Anzai, S.; Masuda, M.; Kohno, S.; Levi, M.; Shiozaki, Y.; Keenan, A.L.; Miyazaki, M. Simultaneous inhibition of FXR and TGR5 exacerbates atherosclerotic formation. J. Lipid Res. 2018, 59, 1709–1713. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.-B.; Liu, X.-Y.; Zhan, W. Farnesoid X receptor agonist INT-767 attenuates liver steatosis and inflammation in rat model of nonalcoholic steatohepatitis. Drug Des. Dev. Ther. 2018, 12, 2213. [Google Scholar] [CrossRef] [Green Version]
- de Luca, C.; Olefsky, J.M. Inflammation and insulin resistance. FEBS Lett. 2008, 582, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Tang, H.; Zhang, C.; Zhao, Y.; Derrien, M.; Rocher, E.; van-Hylckama Vlieg, J.E.; Strissel, K.; Zhao, L.; Obin, M.; et al. Modulation of gut microbiota during probiotic-mediated attenuation of metabolic syndrome in high fat diet-fed mice. ISME J. 2015, 9, 1–15. [Google Scholar] [CrossRef]
- Müller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [CrossRef]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Yadav, H.; Lee, J.H.; Lloyd, J.; Walter, P.; Rane, S.G. Beneficial metabolic effects of a probiotic via butyrate-induced GLP-1 hormone secretion. J. Biol. Chem. 2013, 288, 25088–25097. [Google Scholar] [CrossRef] [Green Version]
- Ducastel, S.; Touche, V.; Trabelsi, M.-S.; Boulinguiez, A.; Butruille, L.; Nawrot, M.; Peschard, S.; Chávez-Talavera, O.; Dorchies, E.; Vallez, E. The nuclear receptor FXR inhibits Glucagon-Like Peptide-1 secretion in response to microbiota-derived Short-Chain Fatty Acids. Sci. Rep. 2020, 10, 1–10. [Google Scholar]
- Pluznick, J. A novel SCFA receptor, the microbiota, and blood pressure regulation. Gut Microbes 2014, 5, 202–207. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, C.B.; Gabe, M.B.N.; Svendsen, B.; Dragsted, L.O.; Rosenkilde, M.M.; Holst, J.J. The impact of short-chain fatty acids on GLP-1 and PYY secretion from the isolated perfused rat colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G53–G65. [Google Scholar] [CrossRef] [Green Version]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotsman, I.; Sharpe, A.H.; Lichtman, A.H. T-cell costimulation and coinhibition in atherosclerosis. Circ. Res. 2008, 103, 1220–1231. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753. [Google Scholar] [CrossRef] [PubMed]
- Siavoshian, S.; Blottiere, H.; Bentouimou, N.; Cherbut, C.; Galmiche, J. Butyrate enhances major histocompatibility complex class I, HLA-DR and ICAM-1 antigen expression on differentiated human intestinal epithelial cells. Eur. J. Clin. Investig. 1996, 26, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Wigren, M.; Rattik, S.; Yao Mattisson, I.; Tomas, L.; Grönberg, C.; Söderberg, I.; Alm, R.; Sundius, L.; Ljungcrantz, I.; Björkbacka, H. Lack of ability to present antigens on major histocompatibility complex class II molecules aggravates atherosclerosis in ApoE−/− mice. Circulation 2019, 139, 2554–2566. [Google Scholar] [CrossRef]
- Neupane, R.; Jin, X.; Sasaki, T.; Li, X.; Murohara, T.; Cheng, X.W. Immune Disorder in Atherosclerotic Cardiovascular Disease―Clinical Implications of Using Circulating T-Cell Subsets as Biomarkers―. Circ. J. 2019, 83, 1431–1438. [Google Scholar] [CrossRef] [Green Version]
- Steffens, S.; Mach, F. Drug Insight: Immunomodulatory effects of statins—Potential benefits for renal patients? Nat. Clin. Pract. Nephrol. 2006, 2, 378–387. [Google Scholar] [CrossRef]
- Jacobsson, L.T.; Turesson, C.; Gülfe, A.; Kapetanovic, M.C.; Petersson, I.F.; Saxne, T.; Geborek, P. Treatment with tumor necrosis factor blockers is associated with a lower incidence of first cardiovascular events in patients with rheumatoid arthritis. J. Rheumatol. 2005, 32, 1213–1218. [Google Scholar]
- Hansson, G.K.; Nilsson, J. Vaccination against atherosclerosis? Induction of atheroprotective immunity. Semin. Immunopathol. 2009, 31, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Mäkinen, P.; Ruotsalainen, A.K.; Ylä-Herttuala, S. Nucleic Acid-Based Therapies for Atherosclerosis. Curr. Atheroscler. Rep. 2020, 22, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arsenault, B.J. The promise and challenges of RNA-targeted therapeutics in preventive cardiology. Eur. Heart J. 2021, ehab462. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoo, J.Y.; Sniffen, S.; McGill Percy, K.C.; Pallaval, V.B.; Chidipi, B. Gut Dysbiosis and Immune System in Atherosclerotic Cardiovascular Disease (ACVD). Microorganisms 2022, 10, 108. https://doi.org/10.3390/microorganisms10010108
Yoo JY, Sniffen S, McGill Percy KC, Pallaval VB, Chidipi B. Gut Dysbiosis and Immune System in Atherosclerotic Cardiovascular Disease (ACVD). Microorganisms. 2022; 10(1):108. https://doi.org/10.3390/microorganisms10010108
Chicago/Turabian StyleYoo, Ji Youn, Sarah Sniffen, Kyle Craig McGill Percy, Veera Bramhachari Pallaval, and Bojjibabu Chidipi. 2022. "Gut Dysbiosis and Immune System in Atherosclerotic Cardiovascular Disease (ACVD)" Microorganisms 10, no. 1: 108. https://doi.org/10.3390/microorganisms10010108
APA StyleYoo, J. Y., Sniffen, S., McGill Percy, K. C., Pallaval, V. B., & Chidipi, B. (2022). Gut Dysbiosis and Immune System in Atherosclerotic Cardiovascular Disease (ACVD). Microorganisms, 10(1), 108. https://doi.org/10.3390/microorganisms10010108