

Pathways for Understanding Blue Carbon Microbiomes with Amplicon Sequencing

 ,
,  , ,
, ,
Abstract
:1. Introduction—Key Knowledge Gaps Amenable to a Blue Carbon Microbiome Meta-Analysis
2. Materials and Methods—Current Data Availability
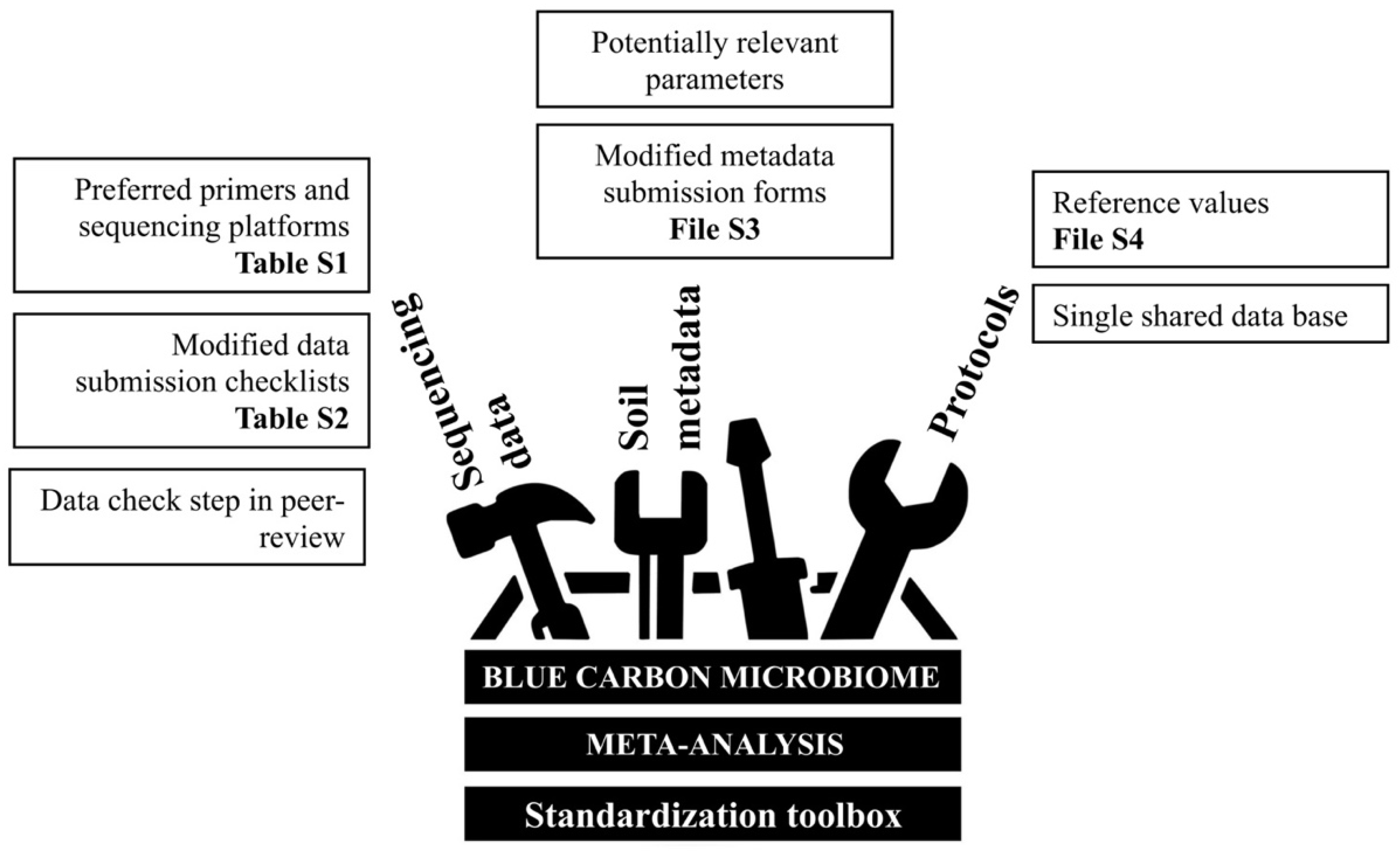
3. Results—Opportunities for Blue Carbon Microbiomes through a Standardisation Toolbox
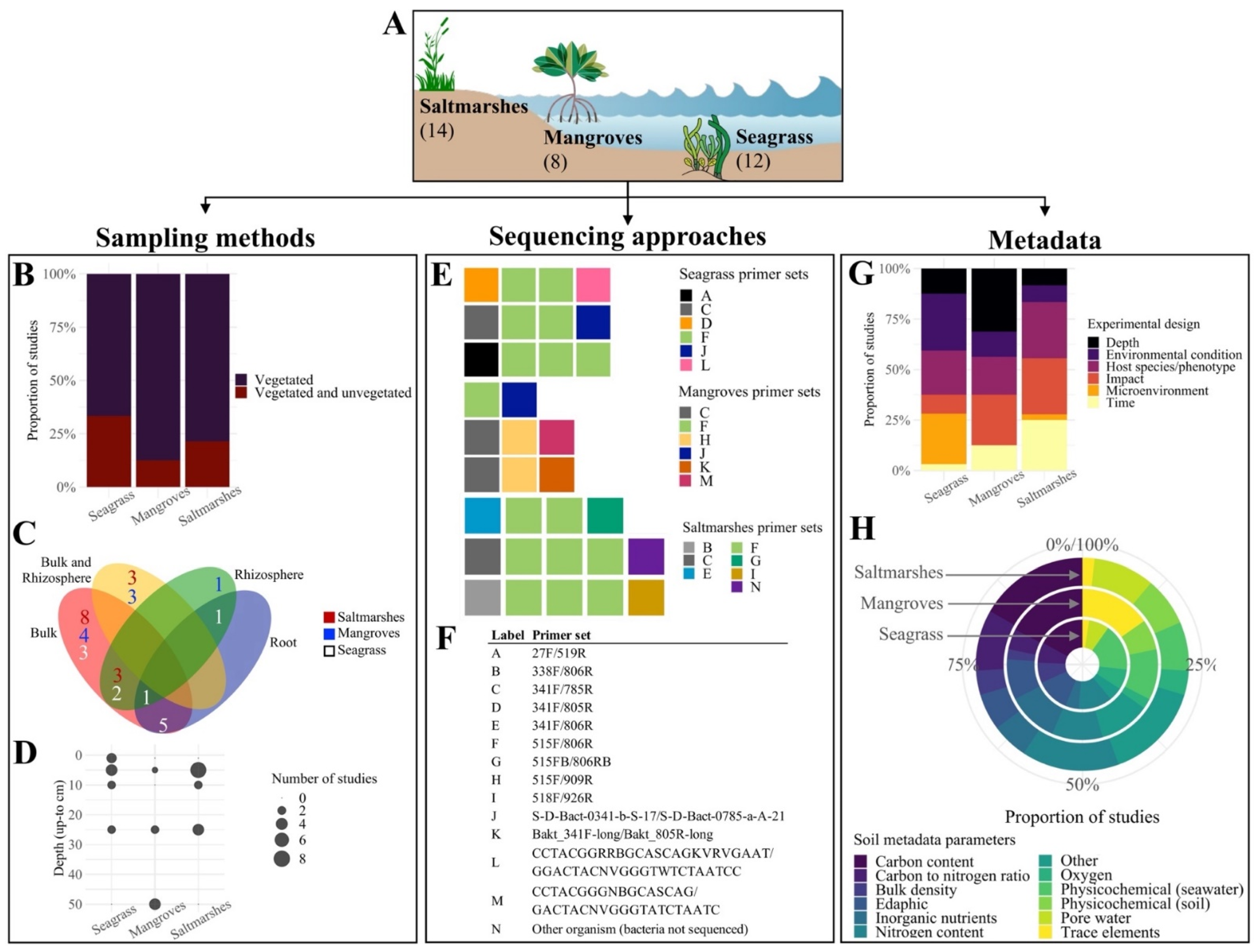
3.1. Sequencing Data
3.2. Soil Metadata and Experimental Designs
3.3. Protocols
4. Discussion—Recommendations and Conclusions
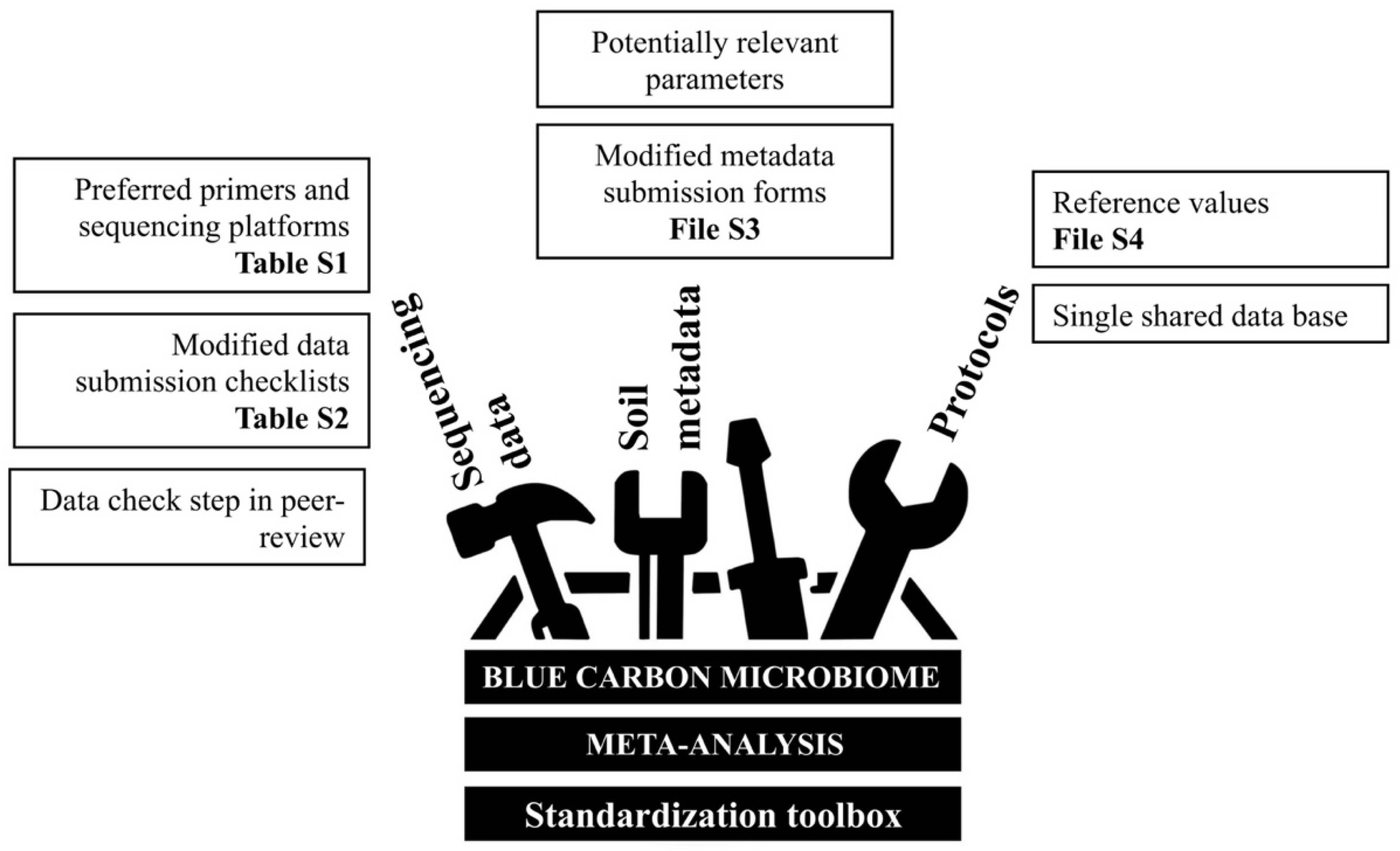
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schallenberg, M.; Kalff, J. The ecology of sediment bacteria in lakes and comparisons with other aquatic ecosystems. Ecology 1993, 74, 919–934. [Google Scholar] [CrossRef]
- Stanier, R.Y. Studies on marine agar-digesting bacteria. J. Bacteriol. 1941, 42, 527–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waksman, S.A.; Carey, C.L.; Reuszer, H.W. Marine bacteria and their role in the cycle of life in the sea: I. Decomposition of marine plant and animal residues by bacteria. Biol. Bull. 1933, 65, 57–79. [Google Scholar] [CrossRef]
- ZoBell, C.E.; Feltham, C.B. The bacterial flora of a marine mud flat as an ecological factor. Ecology 1942, 23, 69–78. [Google Scholar] [CrossRef]
- Ducklow, H. Bacterial production and biomass in the oceans. In Microbial Ecology of the Oceans; Kirchman, D., Ed.; John Wiley and Sons: New York, NY, USA, 2000; Volume 1, pp. 85–120. [Google Scholar]
- Boschker, H.T.S.; Middelburg, J.J. Stable isotopes and biomarkers in microbial ecology. FEMS Microbiol. Ecol. 2002, 40, 85–95. [Google Scholar] [CrossRef]
- Lassen, C.; Ploug, H.; Jørgensen, B.B. A fibre-optic scalar irradiance microsensor: Application for spectral light measurements in sediments. FEMS Microbiol. Lett. 1992, 86, 247–254. [Google Scholar] [CrossRef]
- Pedersen, O.; Revsbech, N.P.; Shabala, S. Microsensors in plant biology: In vivo visualization of inorganic analytes with high spatial and/or temporal resolution. J. Exp. Bot. 2020, 71, 3941–3954. [Google Scholar] [CrossRef]
- Alongi, D.M. The role of bacteria in nutrient recycling in tropical mangrove and other coastal benthic ecosystems. Hydrobiologia 1994, 285, 19–32. [Google Scholar] [CrossRef]
- Boschker, H.T.S.; Wielemaker, A.; Schaub, B.E.M.; Holmer, M. Limited coupling of macrophyte production and bacterial carbon cycling in the sediments of Zostera spp. meadows. Mar. Ecol. Prog. Ser. 2000, 203, 181–189. [Google Scholar] [CrossRef] [Green Version]
- DeLaune, R.D.; Gambrell, R.P.; Pardue, J.H.; Patrick, W.H. Fate of petroleum hydrocarbons and toxic organics in Louisiana coastal environments. Estuaries 1990, 13, 72–80. [Google Scholar] [CrossRef]
- Arnosti, C. Microbial extracellular enzymes and the marine carbon cycle. Annu. Rev. Mar. Sci. 2011, 3, 401–425. [Google Scholar] [CrossRef]
- Spivak, A.C.; Sanderman, J.; Bowen, J.L.; Canuel, E.A.; Hopkinson, C.S. Global-change controls on soil-carbon accumulation and loss in coastal vegetated ecosystems. Nat. Geosci. 2019, 12, 685–692. [Google Scholar] [CrossRef]
- Bahram, M.; Hildebrand, F.; Forslund, S.K.; Anderson, J.L.; Soudzilovskaia, N.A.; Bodegom, P.M.; Bengtsson-Palme, J.; Anslan, S.; Coelho, L.P.; Harend, H.; et al. Structure and function of the global topsoil microbiome. Nature 2018, 560, 233–237. [Google Scholar] [CrossRef] [Green Version]
- Bech, P.K.; Lysdal, K.L.; Gram, L.; Bentzon-Tilia, M.; Strube, M.L. Marine sediments hold an untapped potential for novel taxonomic and bioactive bacterial diversity. mSystems 2020, 5, e00782-20. [Google Scholar] [CrossRef]
- Jochum, L.M.; Schreiber, L.; Marshall, I.P.G.; Jørgensen, B.B.; Schramm, A.; Kjeldsen, K.U. Single-cell genomics reveals a diverse metabolic potential of uncultivated Desulfatiglans-related Deltaproteobacteria widely distributed in marine sediment. Front. Microbiol. 2018, 9, 2038. [Google Scholar] [CrossRef] [Green Version]
- Banister, R.B.; Schwarz, M.T.; Fine, M.; Ritchie, K.B.; Muller, E.M. Instability and stasis among the microbiome of seagrass leaves, roots and rhizomes, and nearby sediments within a natural pH gradient. Microb. Ecol. 2021. [Google Scholar] [CrossRef]
- Cúcio, C.; Engelen, A.H.; Costa, R.; Muyzer, G. Rhizosphere microbiomes of European seagrasses are selected by the plant, but are not species specific. Front. Microbiol. 2016, 7, 440. [Google Scholar] [CrossRef] [Green Version]
- Fahimipour, A.K.; Kardish, M.R.; Lang, J.M.; Green, J.L.; Eisen, J.A.; Stachowicz, J.J. Global-scale structure of the eelgrass microbiome. Appl. Environ. Microbiol. 2017, 83, e03391-16. [Google Scholar] [CrossRef] [Green Version]
- Hurtado-McCormick, V.; Kahlke, T.; Petrou, K.; Jeffries, T.; Ralph, P.J.; Seymour, J.R. Regional and microenvironmental scale characterization of the Zostera muelleri seagrass microbiome. Front. Microbiol. 2019, 10, 1011. [Google Scholar] [CrossRef]
- Hanley, T.C.; Bowen, J.L.; Kearns, P.J.; Hughes, A.R. Short-and long-term effects of nutrient enrichment on salt marsh plant production and microbial community structure. J. Ecol. 2021, 109, 3779–3793. [Google Scholar] [CrossRef]
- Kolton, M.; Rolando, J.L.; Kostka, J.E. Elucidation of the rhizosphere microbiome linked to Spartina alterniflora phenotype in a salt marsh on Skidaway Island, Georgia, USA. FEMS Microbiol. Ecol. 2020, 96, fiaa026. [Google Scholar] [CrossRef]
- Martin, B.C.; Middleton, J.A.; Skrzypek, G.; Kendrick, G.A.; Cosgrove, J.; Fraser, M.W. Composition of seagrass root associated bacterial communities are linked to nutrients and heavy metal concentrations in an anthropogenically influenced estuary. Front. Mar. Sci. 2022, 8, 768864. [Google Scholar] [CrossRef]
- Nóbrega, M.S.; Silva, B.S.; Tschoeke, D.A.; Appolinario, L.R.; Calegario, G.; Venas, T.M.; Macedo, L.; Asp, N.; Cherene, B.; Marques, J.S.J.; et al. Mangrove microbiome reveals importance of sulfur metabolism in tropical coastal waters. Sci. Total Environ. 2022, 813, 151889. [Google Scholar] [CrossRef]
- Ren, L.; Jensen, K.; Porada, P.; Mueller, P. Biota-mediated carbon cycling—A synthesis of biotic-interaction controls on blue carbon. Ecol. Lett. 2022, 25, 521–540. [Google Scholar] [CrossRef]
- Serrano, O.; Lavery, P.S.; Duarte, C.M.; Kendrick, G.A.; Calafat, A.; York, P.H.; Steven, A.; Macreadie, P.I. Can mud (silt and clay) concentration be used to predict soil organic carbon content within seagrass ecosystems? Biogeosciences 2016, 13, 4915–4926. [Google Scholar] [CrossRef] [Green Version]
- Kallenbach, C.M.; Frey, S.D.; Grandy, A.S. Direct evidence for microbial-derived soil organic matter formation and its ecophysiological controls. Nat. Commun. 2016, 7, 13630. [Google Scholar] [CrossRef] [Green Version]
- Allard, S.M.; Costa, M.T.; Bulseco, A.N.; Helfer, V.; Wilkins, L.G.E.; Hassenrück, C.; Zengler, K.; Zimmer, M.; Erazo, N.; Mazza Rodrigues, J.L.; et al. Introducing the mangrove microbiome initiative: Identifying microbial research priorities and approaches to better understand, protect, and rehabilitate mangrove ecosystems. mSystems 2020, 5, e00658-20. [Google Scholar] [CrossRef]
- Trevathan-Tackett, S.M.; Sherman, C.D.H.; Huggett, M.J.; Campbell, A.H.; Laverock, B.; Hurtado-McCormick, V.; Seymour, J.R.; Firl, A.; Messer, L.F.; Ainsworth, T.D.; et al. A horizon scan of priorities for coastal marine microbiome research. Nat. Ecol. Evol. 2019, 3, 1509–1520. [Google Scholar] [CrossRef]
- Howard, J.; Hoyt, S.; Isensee, K.; Pidgeon, E.; Telszewski, M. Coastal Blue Carbon: Methods for Assessing Carbon Stocks and Emissions Factors in Mangroves, Tidal Salt Marshes, and Seagrass Meadows; Conservation International, Intergovernmental Oceanographic Commission of UNESCO, International Union for Conservation of Nature: Arlington, VA, USA, 2014. [Google Scholar]
- Liu, S.; Trevathan-Tackett, S.M.; Jiang, Z.; Cui, L.; Wu, Y.; Zhang, X.; Li, J.; Luo, H.; Huang, X. Nutrient loading decreases blue carbon by mediating fungi activities within seagrass meadows. Environ. Res. 2022, 212, 113280. [Google Scholar] [CrossRef]
- Kearns, P.J.; Bulseco-McKim, A.N.; Hoyt, H.; Angell, J.H.; Bowen, J.L. Nutrient enrichment alters salt marsh fungal communities and promotes putative fungal denitrifiers. Microb. Ecol. 2019, 77, 358–369. [Google Scholar] [CrossRef]
- Luis, P.; Saint-Genis, G.; Vallon, L.; Bourgeois, C.; Bruto, M.; Marchand, C.; Record, E.; Hugoni, M. Contrasted ecological niches shape fungal and prokaryotic community structure in mangroves sediments. Environ. Microbiol. 2019, 21, 1407–1424. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, P.; Kottmann, R.; Field, D.; Knight, R.; Cole, J.R.; Amaral-Zettler, L.; Gilbert, J.A.; Karsch-Mizrachi, I.; Johnston, A.; Cochrane, G.; et al. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat. Biotechnol. 2011, 29, 415–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.A.; Díez-Vives, C.; Majzoub, M.E.; Nielsen, S.; Thomas, T. Stress response of the marine sponge Scopalina sp. Can microbial community composition predict sponge disease? FEMS Microbiol. Ecol. 2021, 97, fiab095. [Google Scholar] [CrossRef] [PubMed]
- Rosado, P.M.; Leite, D.C.A.; Duarte, G.A.S.; Chaloub, R.M.; Jospin, G.; da Rocha, U.N.; Saraiva, J.P.; Dini-Andreote, F.; Eisen, J.A.; Bourne, D.G.; et al. Marine probiotics: Increasing coral resistance to bleaching through microbiome manipulation. ISME J. 2019, 13, 921–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Ye, Q.; Sanders, C.J.; Du, J.; Zhang, J. Bacterial-derived nutrient and carbon source-sink behaviors in a sandy beach subterranean estuary. Mar. Pollut. Bull. 2020, 160, 111570. [Google Scholar] [CrossRef]
- Classen, A.T.; Sundqvist, M.K.; Henning, J.A.; Newman, G.S.; Moore, J.A.M.; Cregger, M.A.; Moorhead, L.C.; Patterson, C.M. Direct and indirect effects of climate change on soil microbial and soil microbial-plant interactions: What lies ahead? Ecosphere 2015, 6, 130. [Google Scholar] [CrossRef]
- Mueller, P.; Granse, D.; Nolte, S.; Weingartner, M.; Hoth, S.; Jensen, K. Unrecognized controls on microbial functioning in Blue Carbon ecosystems: The role of mineral enzyme stabilization and allochthonous substrate supply. Ecol. Evol. 2020, 10, 998–1011. [Google Scholar] [CrossRef] [Green Version]
- Aires, T.; Stuij, T.M.; Muyzer, G.; Serrão, E.A.; Engelen, A.H. Characterization and comparison of bacterial communities of an invasive and two native Caribbean seagrass species sheds light on the possible influence of the microbiome on invasive mechanisms. Front. Microbiol. 2021, 12, 653998. [Google Scholar] [CrossRef]
- Kohn, T.; Rast, P.; Kallscheuer, N.; Wiegand, S.; Boedeker, C.; Jetten, M.S.M.; Jeske, O.; Vollmers, J.; Kaster, A.K.; Rohde, M.; et al. The microbiome of Posidonia oceanica seagrass leaves can be dominated by Planctomycetes. Front. Microbiol. 2020, 11, 1458. [Google Scholar] [CrossRef]
- Ceccon, D.M.; Faoro, H.; da Cunha Lana, P.; de Souza, E.M.; de Oliveira Pedrosa, F. Metataxonomic and metagenomic analysis of mangrove microbiomes reveals community patterns driven by salinity and pH gradients in Paranaguá Bay, Brazil. Sci. Total Environ. 2019, 694, 133609. [Google Scholar] [CrossRef]
- Zhuang, W.; Yu, X.; Hu, R.; Luo, Z.; Liu, X.; Zheng, X.; Xiao, F.; Peng, Y.; He, Q.; Tian, Y.; et al. Diversity, function and assembly of mangrove root-associated microbial communities at a continuous fine-scale. Npj Biofilms Microbiomes 2020, 6, 52. [Google Scholar] [CrossRef]
- Lumibao, C.Y.; Bernik, B.M.; Formel, S.K.; Kandalepas, D.; Mighell, K.L.; Pardue, J.; Van Bael, S.A.; Blum, M.J. Rhizosphere microbial communities reflect genotypic and trait variation in a salt marsh ecosystem engineer. Am. J. Bot. 2020, 107, 941–949. [Google Scholar] [CrossRef]
- Mavrodi, O.V.; Jung, C.M.; Eberly, J.O.; Hendry, S.V.; Namjilsuren, S.; Biber, P.D.; Indest, K.J.; Mavrodi, D.V. Rhizosphere microbial communities of Spartina alterniflora and Juncus roemerianus from restored and natural tidal marshes on Deer Island, Mississippi. Front. Microbiol. 2018, 9, 3049. [Google Scholar] [CrossRef]
- Bowen, J.L.; Kearns, P.J.; Byrnes, J.E.; Wigginton, S.; Allen, W.J.; Greenwood, M.; Tran, K.; Yu, J.; Cronin, J.T.; Meyerson, L.A. Lineage overwhelms environmental conditions in determining rhizosphere bacterial community structure in a cosmopolitan invasive plant. Nat. Commun. 2017, 8, 433. [Google Scholar] [CrossRef]
- Kearns, P.J.; Angell, J.H.; Howard, E.M.; Deegan, L.A.; Stanley, R.H.; Bowen, J.L. Nutrient enrichment induces dormancy and decreases diversity of active bacteria in salt marsh sediments. Nat. Commun. 2016, 7, 12881. [Google Scholar] [CrossRef] [Green Version]
- Friesen, S.D.; Dunn, C.; Freeman, C. Decomposition as a regulator of carbon accretion in mangroves: A review. Ecol. Eng. 2018, 114, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Trevathan-Tackett, S.M.; Kepfer-Rojas, S.; Engelen, A.H.; York, P.H.; Ola, A.; Li, J.; Kelleway, J.J.; Jinks, K.I.; Jackson, E.L.; Adame, M.F. Ecosystem type drives tea litter decomposition and associated prokaryotic microbiome communities in freshwater and coastal wetlands at a continental scale. Sci. Total Environ. 2021, 782, 146819. [Google Scholar] [CrossRef]
- Averill, C.; Werbin, Z.R.; Atherton, K.F.; Bhatnagar, J.M.; Dietze, M.C. Soil microbiome predictability increases with spatial and taxonomic scale. Nat. Ecol. Evol. 2021, 5, 747–756. [Google Scholar] [CrossRef]
- Zhang, K.; Delgado-Baquerizo, M.; Zhu, Y.G.; Chu, H. Space is more important than season when shaping soil microbial communities at a large spatial scale. mSystems 2020, 5, e00783-19. [Google Scholar] [CrossRef]
- Martin, B.C.; Gleeson, D.; Statton, J.; Siebers, A.R.; Grierson, P.; Ryan, M.H.; Kendrick, G.A. Low light availability alters root exudation and reduces putative beneficial microorganisms in seagrass roots. Front. Microbiol. 2018, 8, 2667. [Google Scholar] [CrossRef]
- Oliveira De Santana, C.; Spealman, P.; Maciel Melo, V.M.; Gresham, D.; Bomfim de Jesus, T.; Chinalia, F.A. Effects of tidal influence on the structure and function of prokaryotic communities in the sediments of a pristine Brazilian mangrove. Biogeosciences 2021, 18, 2259–2273. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, H.; Song, Z.; Huang, Y.; Hu, X. Seasonal dynamics of Bathyarchaeota-dominated benthic archaeal communities associated with seagrass (zostera japonica) meadows. J. Mar. Sci. Eng. 2021, 9, 1304. [Google Scholar] [CrossRef]
- Tiralerdpanich, P.; Nasaree, S.; Pinyakong, O.; Sonthiphand, P. Variation of the mangrove sediment microbiomes and their phenanthrene biodegradation rates during the dry and wet seasons. Environ. Pollut. 2021, 289, 117849. [Google Scholar] [CrossRef]
- Dini-Andreote, F.; de Cássia Pereira e Silva, M.; Triado-Margarit, X.; Casamayor, E.O.; van Elsas, J.D.; Falcão Salles, J. Dynamics of bacterial community succession in a salt marsh chronosequence: Evidences for temporal niche partitioning. ISME J. 2014, 8, 1989–2001. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Yang, P.; Falcão Salles, J. Distribution of root-associated bacterial communities along a salt-marsh primary succession. Front. Plant Sci. 2016, 6, 1188. [Google Scholar] [CrossRef] [Green Version]
- Macreadie, P.I.; Atwood, T.B.; Seymour, J.R.; Schmitz Fontes, M.L.; Sanderman, J.; Nielsen, D.A.; Connolly, R.M. Vulnerability of seagrass blue carbon to microbial attack following exposure to warming and oxygen. Sci. Total Environ. 2019, 686, 264–275. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Jansson, J.K.; Knight, R. The Earth Microbiome project: Successes and aspirations. BMC Biol. 2014, 12, 69. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, X.; Lee, S.Y. Updated estimates of carbon accumulation rates in coastal marsh sediments. Biogeosciences 2014, 11, 5057–5071. [Google Scholar] [CrossRef] [Green Version]
- Duvallet, C.; Gibbons, S.M.; Gurry, T.; Irizarry, R.A.; Alm, E.J. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat. Commun. 2017, 8, 1784. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Graber, A.; McBurney, R.N.; Balasubramanian, R. Sample size and statistical power considerations in high-dimensionality data settings: A comparative study of classification algorithms. BMC Bioinform. 2010, 11, 447. [Google Scholar] [CrossRef]
- Diepenbroek, M.; Grobe, H.; Reinke, M.; Schindler, U.; Schlitzer, R.; Sieger, R.; Wefer, G. PANGAEA—An information system for environmental sciences. Comput. Geosci. 2002, 28, 1201–1210. [Google Scholar] [CrossRef] [Green Version]
- Gries, C.; Servilla, M.; O’Brien, M.; Vanderbilt, K.; Smith, C.; Costa, D.; Grossman-Clarke, S. Achieving FAIR data principles at the environmental data initiative, the US-LTER data repository. Biodivers. Inf. Sci. Stand. 2019, 3, e37047. [Google Scholar] [CrossRef]
- Bissett, A.; Fitzgerald, A.; Meintjes, T.; Mele, P.M.; Reith, F.; Dennis, P.G.; Breed, M.F.; Brown, B.; Brown, M.V.; Brugger, J.; et al. Introducing BASE: The Biomes of Australian Soil Environments soil microbial diversity database. GigaScience 2016, 5, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulmer, R.H.; Stephenson, F.; Jones, H.F.E.; Townsend, M.; Hillman, J.R.; Schwendenmann, L.; Lundquist, C.J. Blue carbon stocks and cross-habitat subsidies. Front. Mar. Sci. 2020, 7, 380. [Google Scholar] [CrossRef]
- Fest, B.J.; Swearer, S.E.; Arndt, S.K. A review of sediment carbon sampling methods in mangroves and their broader impacts on stock estimates for blue carbon ecosystems. Sci. Total Environ. 2022, 816, 151618. [Google Scholar] [CrossRef]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, J.; Singh, K.; Fern, A.; Kirton, E.S.; He, S.; Woyke, T.; Lee, J.; Chen, F.; Dangl, J.L.; Tringe, S.G. Primer and platform effects on 16S rRNA tag sequencing. Front. Microbiol. 2015, 6, 771. [Google Scholar] [CrossRef] [Green Version]
- Schirmer, M.; Ijaz, U.Z.; D’Amore, R.; Hall, N.; Sloan, W.T.; Quince, C. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 2015, 43, e37. [Google Scholar] [CrossRef] [Green Version]
- Dabney, J.; Meyer, M. Length and GC-biases during sequencing library amplification: A comparison of various polymerase-buffe systems with ancient and modern DNA sequencing libraries. BioTechniques 2012, 52, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Wen, C.; Qin, Y.; Yin, H.; Tu, Q.; Van Nostrand, J.D.; Yuan, T.; Yuan, M.; Deng, Y.; Zhou, J. Phasing amplicon sequencing on Illumina Miseq for robust environmental microbial community analysis. BMC Microbiol. 2015, 15, 125. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Caruana, R.; Niculescu-Mizil, A. An empirical comparison of supervised learning algorithms. In Proceedings of the 23rd International Conference on Machine Learning, Pittsburgh, PA, USA, 25–29 June 2006; pp. 161–168. [Google Scholar]
- Roguet, A.; Eren, A.M.; Newton, R.J.; McLellan, S.L. Fecal source identification using random forest. Microbiome 2018, 6, 185. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaf, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372. [Google Scholar] [CrossRef]
- Hankeln, W.; Buttigieg, P.L.; Fink, D.; Kottmann, R.; Yilmaz, P.; Glöckner, F.O. MetaBar-a tool for consistent contextual data acquisition and standards compliant submission. BMC Bioinform. 2010, 11, 358. [Google Scholar] [CrossRef] [Green Version]
- Kelleway, J.J.; Serrano, O.; Baldock, J.A.; Burgess, R.; Cannard, T.; Lavery, P.S.; Lovelock, C.E.; Macreadie, P.I.; Masqué, P.; Newnham, M.; et al. A national approach to greenhouse gas abatement through blue carbon management. Glob. Environ. Chang. 2020, 63, 102083. [Google Scholar] [CrossRef]
- Zemliansky, P.; St Amant, K. Handbook of Research on Virtual Workplaces and the New Nature of Business Practices; Information Science Rereference: Hershey, NY, USA, 2008. [Google Scholar]
- Jones, M.B.; O’Brien, M.; Mecum, B.; Boettiger, C.; Schildhauer, M.; Maier, M.; Whiteaker, T.; Earl, S.; Chong, S. Ecological Metadata Language (EML) Version 2.2; KNB Data Repository, 2019; Available online: https://eml.ecoinformatics.org/ (accessed on 17 March 2022).

{kind=link}
{kind=link}
{kind=link}
| Research Question | Meta-Analysis Approach | Methodological Constraints | Result | Technical Issues | Previously Proposed Solutions | Additional Solutions (This Study) | Supplementary Materials |
|---|---|---|---|---|---|---|---|
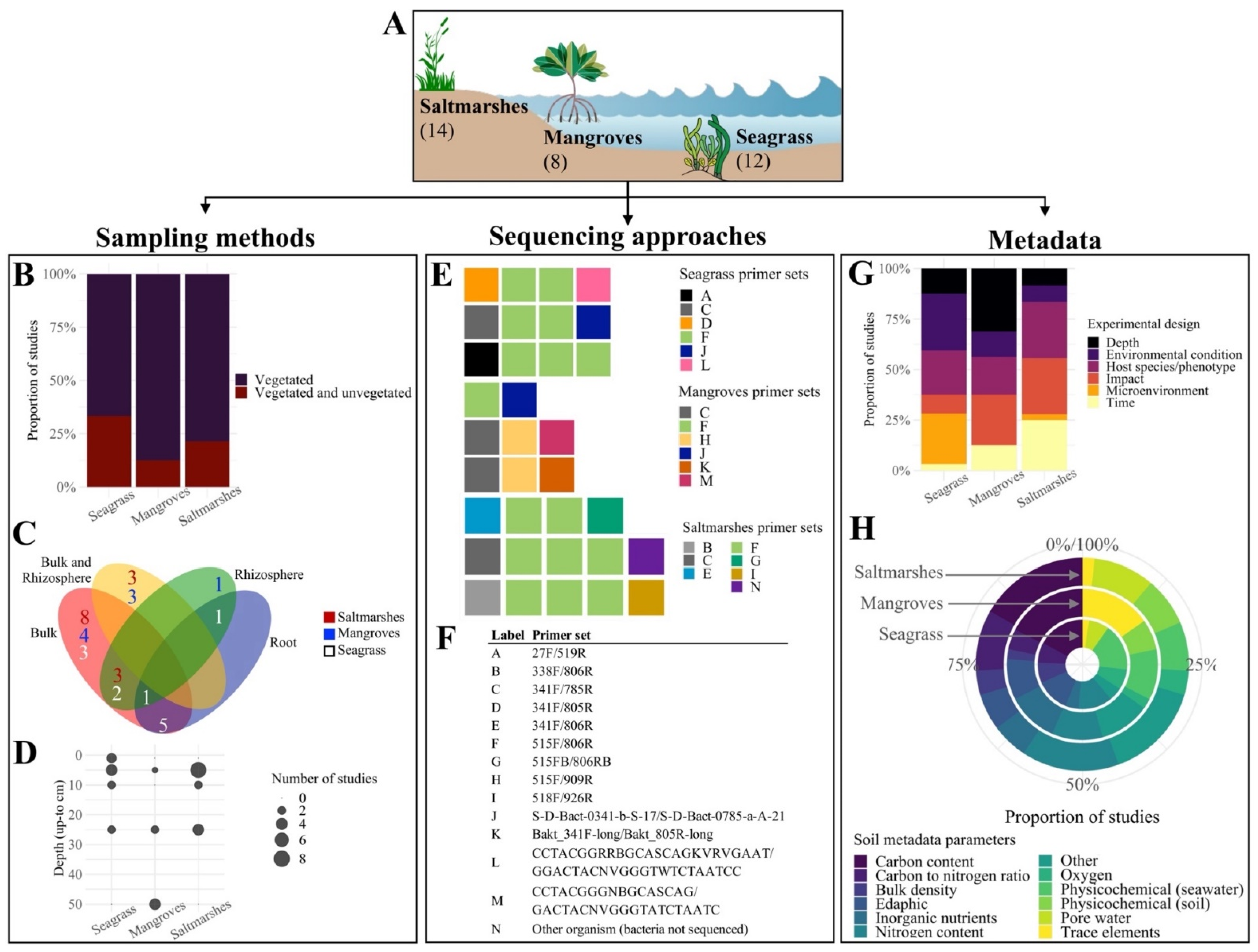
| Is there a Blue Carbon soil microbiome or a shared “Blue Carbon microbial signature” between BCEs? | Combine multiple studies from seagrass, mangroves, and saltmarshes | Variable sequencing approaches used to generate data from BCEs | Prevents comparisons between datasets through data pooling | Inconsistent primer sets | MIMARKS [34] | Preferred primer sets and sequencing platforms | Primers and sequencing platforms list (Table S1) |
| Sample index or mapping file accessibility | Established minimal requirements for SRA/EBI-ENA submissions or alike | Modified submission checklists, including mandatory tabs for data format and sample ID. Example with MIMARKS [34] | Checklist modifications (Table S2) | ||||
| Not-optimal sequencing data formats | |||||||
| Missing sequencing files or samples in mapping files submitted to data repositories | Implemented data curation in peer-review | Data check step in peer-review; i.e., production editor to assure the submission of complete datasets to the repositories | Not applicable | ||||
| Is the Blue Carbon microbiome linked to soil carbon content and other Blue Carbon soil metrics? | Run separate random forest classifiers within studies that measure soil carbon density | Normalised carbon density data often not measured | Limited normalised carbon density data, which require measurements of both percent of organic carbon and dry bulk density | Carbon density data not collected | Research focus on resolving finer cause-–effect and correlative details surrounding the microbiome | Latest advances on the topic, with suggested potentially relevant parameters | Not applicable |
| Raw data not available (only graphical summaries/averages published) | Global database with parameters set up; i.e., targeted carbon data repositories [63,64] | Modified metadata submission form with mandatory fields. Example with EDI [64] | Form modifications (File S3) | ||||
| Invitation for everyone to contribute collaboratively, marketing campaigns | Not applicable | ||||||
| What is the effect of other environmental and edaphic parameters on the Blue Carbon microbiome? | Include multiple environmental parameters as factors | Varying soil metadata, often specific to the treatments or hypotheses of each study | Variable parameters for soil metadata, measured at differing depths | Multiple parameters to inform on carbon content | Suggested standards from global initiatives; e.g., EMP [59], BASE [65]. | Suggested reference values. Example using Blue Carbon Manual worksheet [30] | Reference values worksheet (File S4) |
| Multiple units for the same parameter | Proposal of a single shared database for established standard methods, protocols, and reference values | Not applicable | |||||
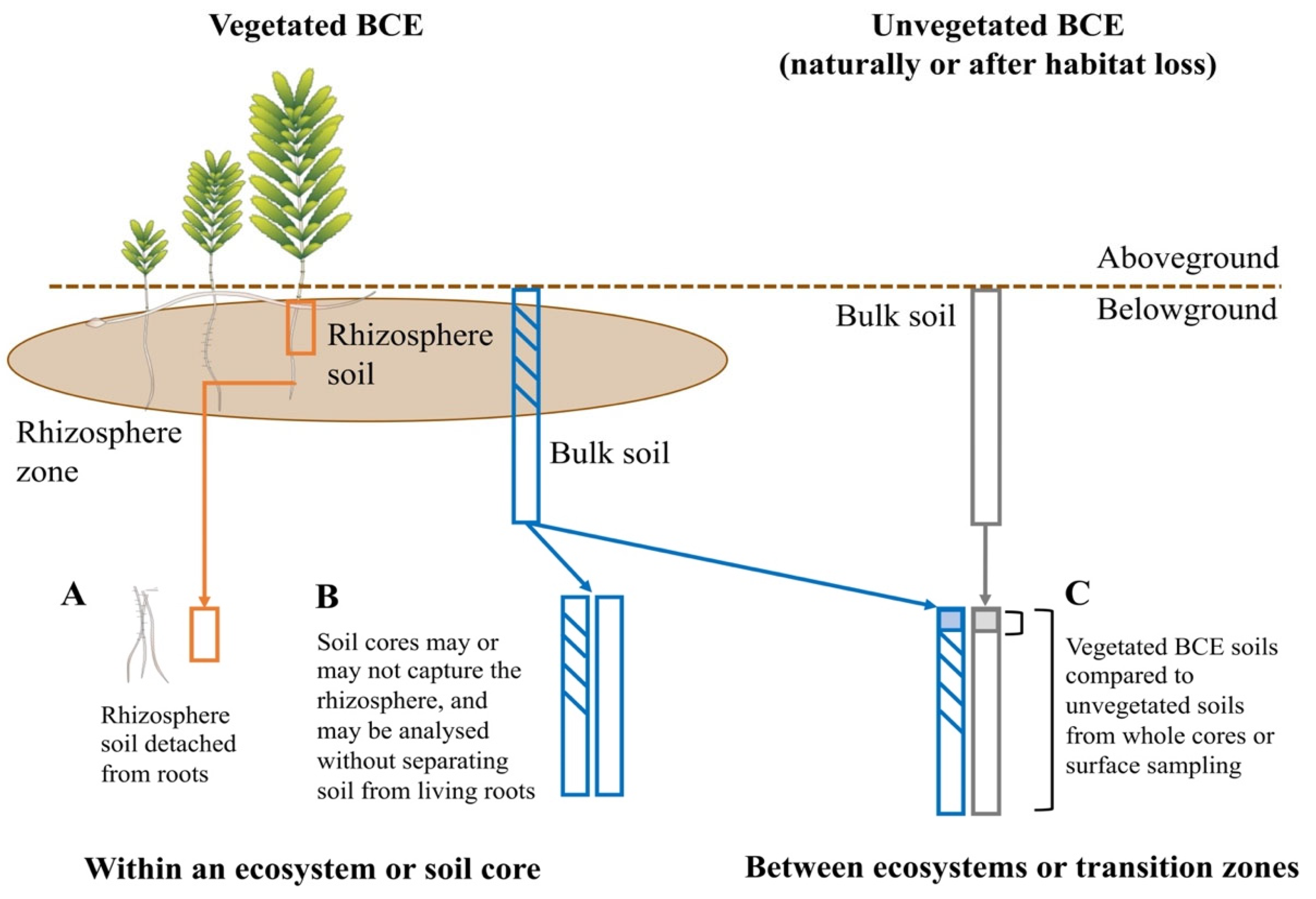
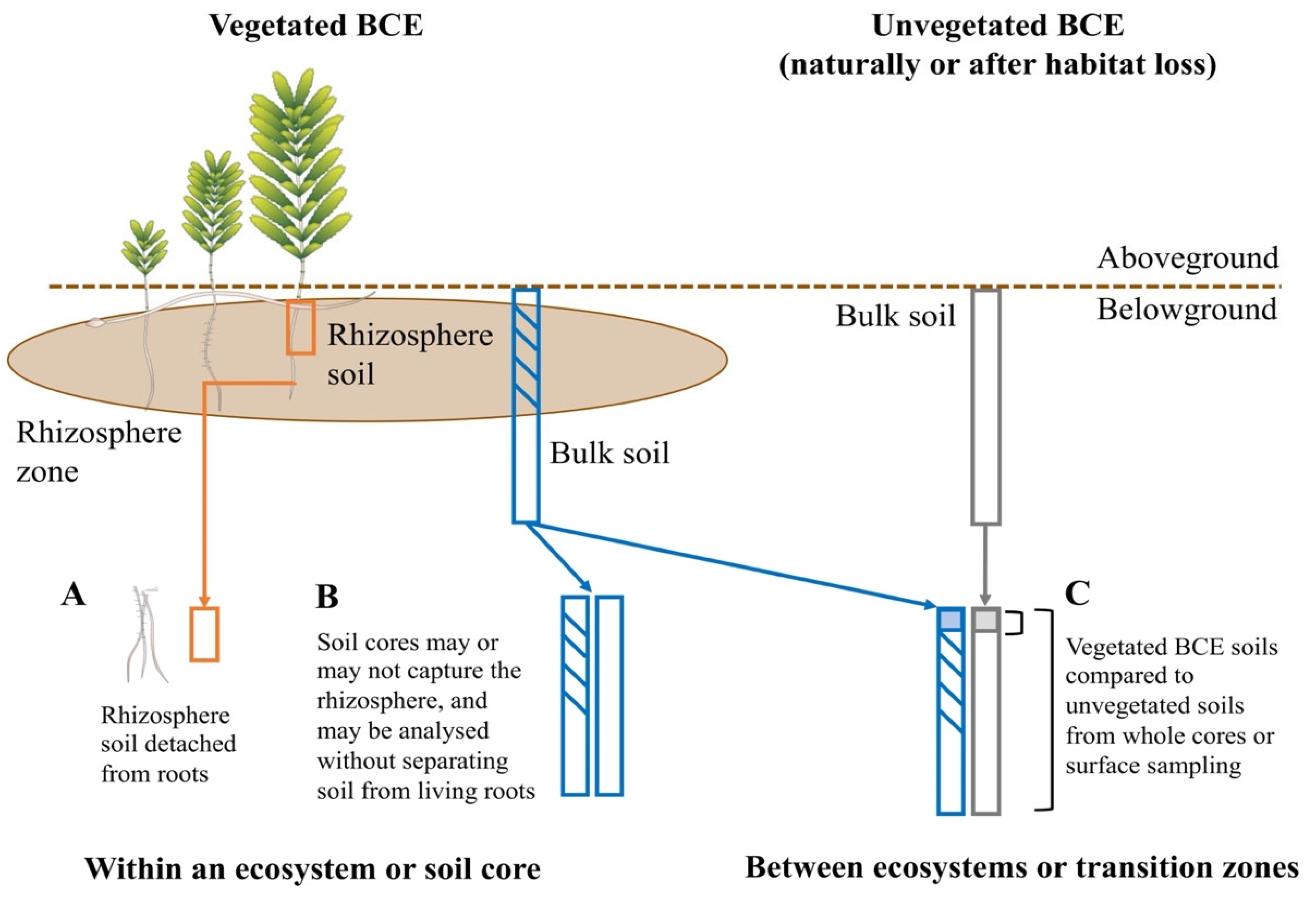
| Do inter- and intra-specific variation influence soil microbiomes in BCEs? | Example with vegetation type: run random forest classifiers within studies that predict habitat | Few studies with required experimental design; i.e., with vegetated and unvegetated samples collected at the same depth | Reduced statistical power of classification algorithms | Experimental designs influenced by within-ecosystem research interests | Design and implementation of studies to understand influence of vegetation and cross-habitat subsidies of carbon on microbiomes and soil parameters [66] | Not applicable | Not applicable |
| Would this Blue Carbon signature change across different spatio-temporal scales? | Include studies from different biogeographical locations and seasons | Current microbiome studies influenced by research interests or funds availability | Lack of studies with experimental designs aligned with the key research questions of this study | Not applicable | Not applicable | Not applicable | Not applicable |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hurtado-McCormick, V.; Trevathan-Tackett, S.M.; Bowen, J.L.; Connolly, R.M.; Duarte, C.M.; Macreadie, P.I. Pathways for Understanding Blue Carbon Microbiomes with Amplicon Sequencing. Microorganisms 2022, 10, 2121. https://doi.org/10.3390/microorganisms10112121
Hurtado-McCormick V, Trevathan-Tackett SM, Bowen JL, Connolly RM, Duarte CM, Macreadie PI. Pathways for Understanding Blue Carbon Microbiomes with Amplicon Sequencing. Microorganisms. 2022; 10(11):2121. https://doi.org/10.3390/microorganisms10112121
Chicago/Turabian StyleHurtado-McCormick, Valentina, Stacey M. Trevathan-Tackett, Jennifer L. Bowen, Rod M. Connolly, Carlos M. Duarte, and Peter I. Macreadie. 2022. "Pathways for Understanding Blue Carbon Microbiomes with Amplicon Sequencing" Microorganisms 10, no. 11: 2121. https://doi.org/10.3390/microorganisms10112121
APA StyleHurtado-McCormick, V., Trevathan-Tackett, S. M., Bowen, J. L., Connolly, R. M., Duarte, C. M., & Macreadie, P. I. (2022). Pathways for Understanding Blue Carbon Microbiomes with Amplicon Sequencing. Microorganisms, 10(11), 2121. https://doi.org/10.3390/microorganisms10112121