
Effects of Preservation and Propagation Methodology on Microcosms Derived from the Oral Microbiome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Background
2. Methods
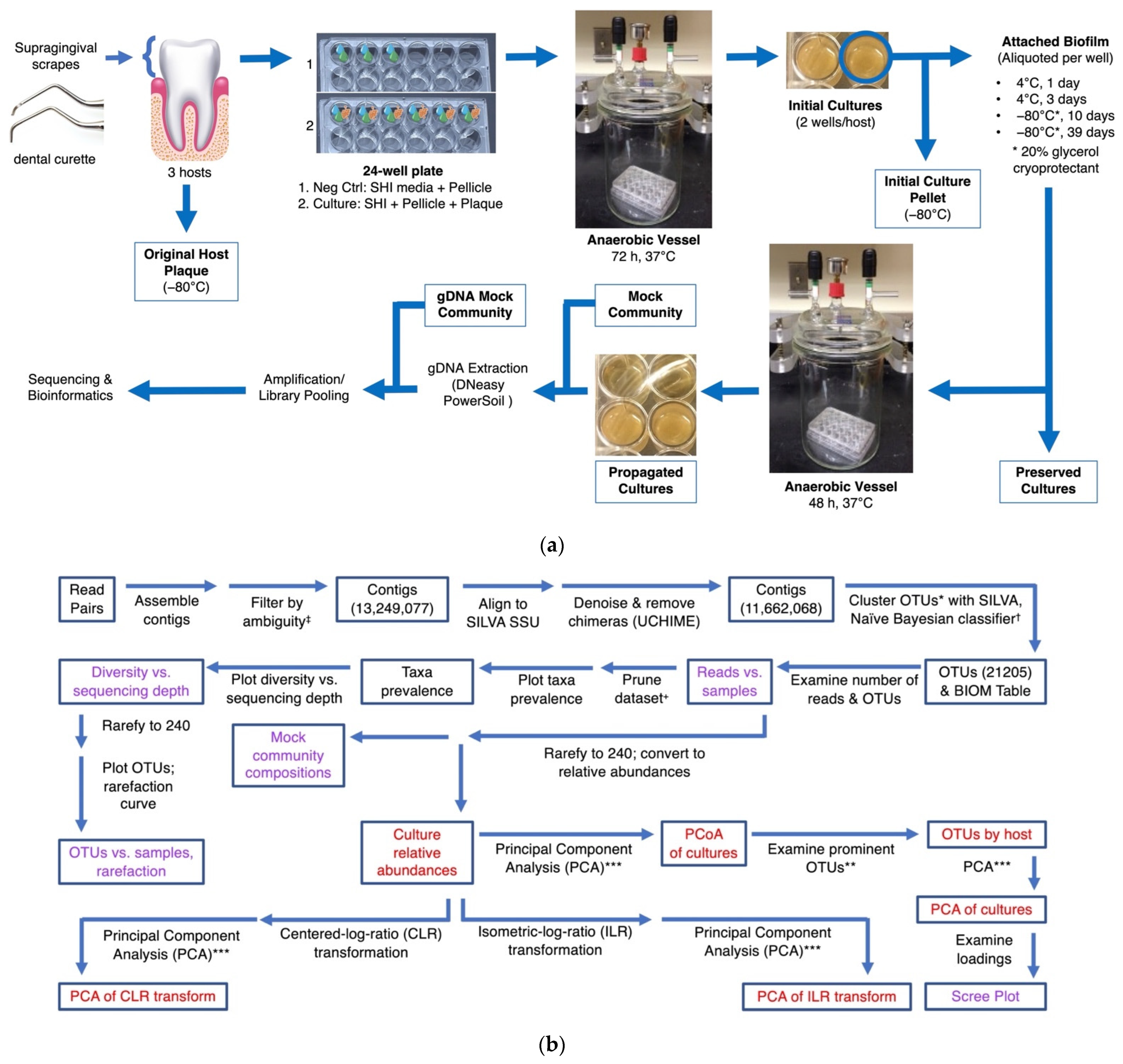
2.1. Sample Collection and Culturing of Human Plaque Communities
2.2. DNA Extraction
2.3. Amplicon Library Construction and Illumina MiSeq Sequencing
2.4. 16S rRNA Bioinformatics Analysis
2.5. Statistical Analysis
3. Results
3.1. Sample Collection, Controls, and Rarefaction
3.2. Decrease in Community Diversity from Initial Plaque Culturing
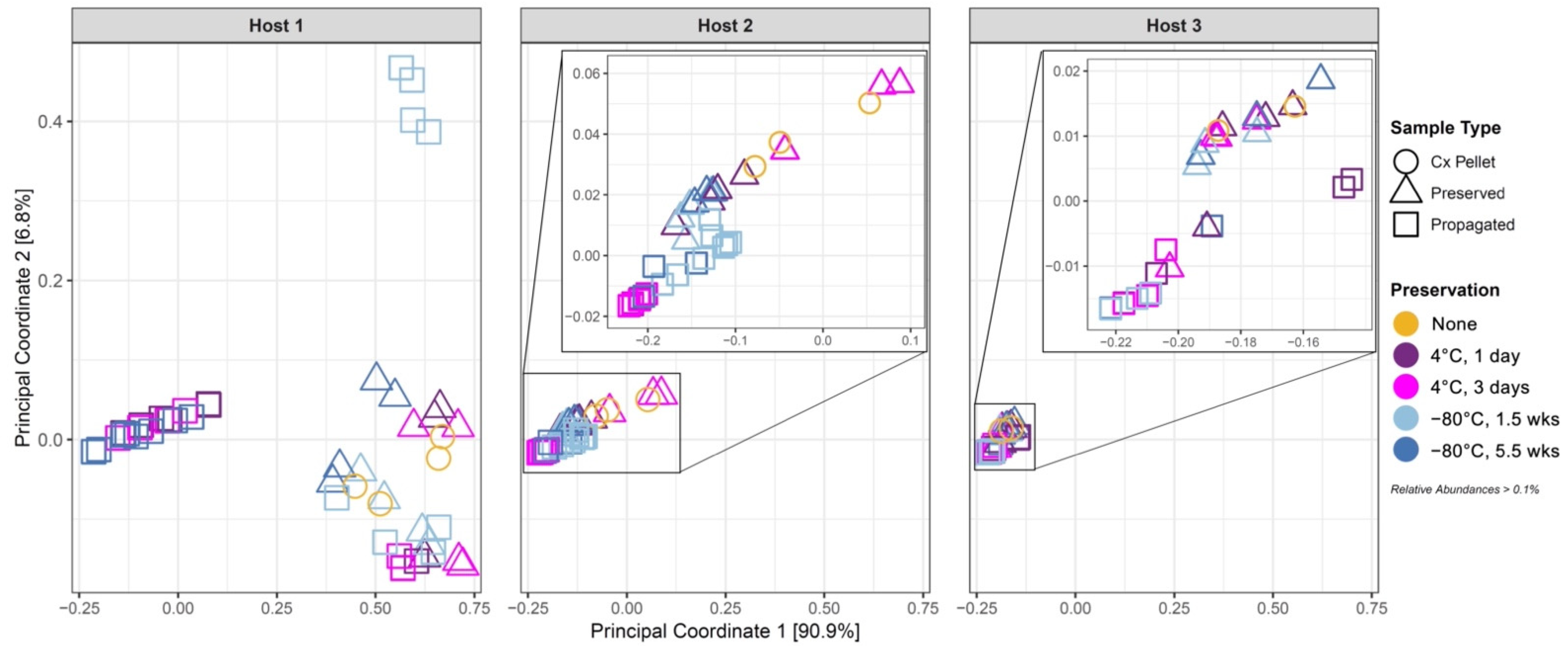
3.3. Effect of Cold Preservation on Community Composition
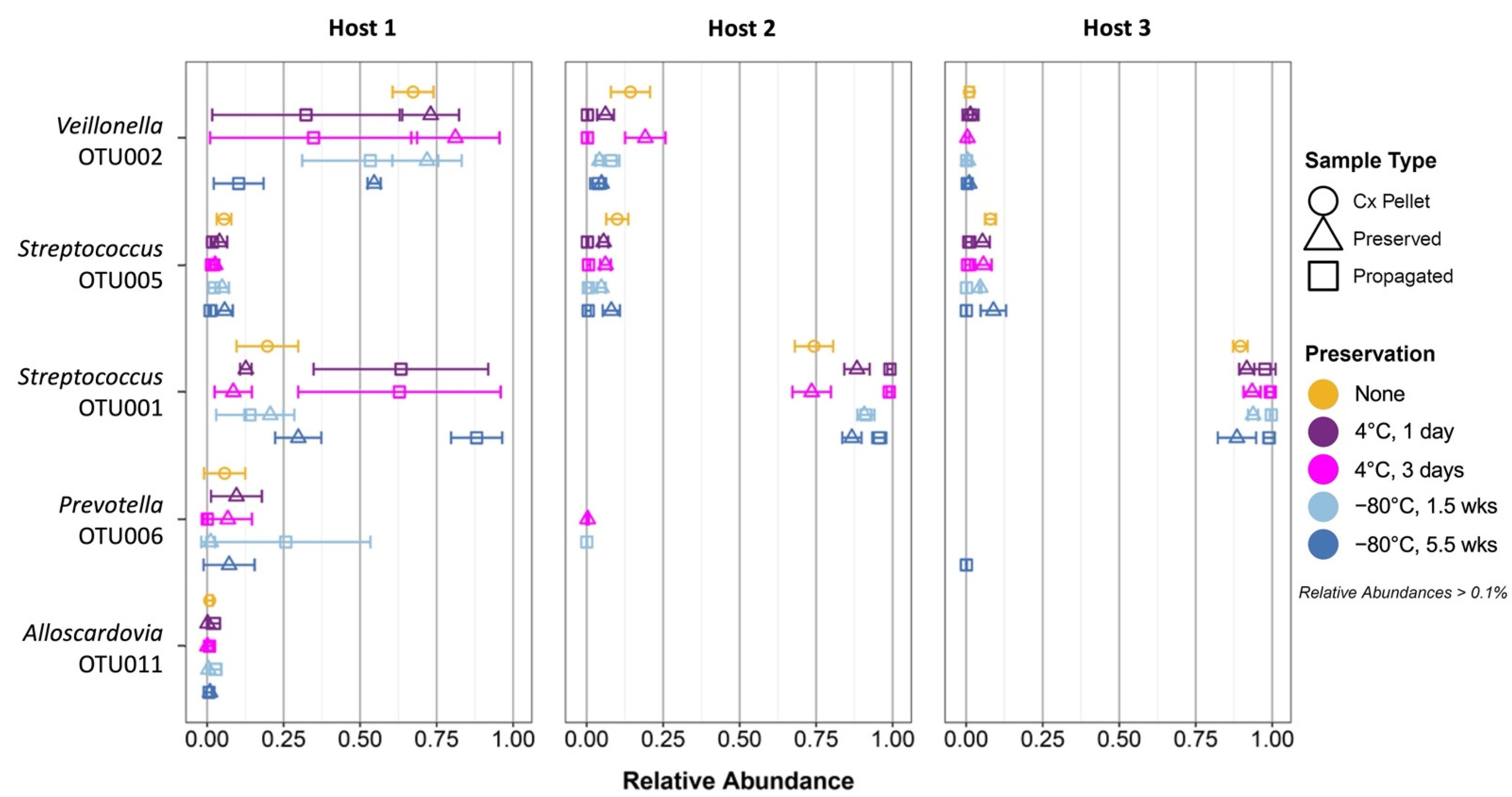
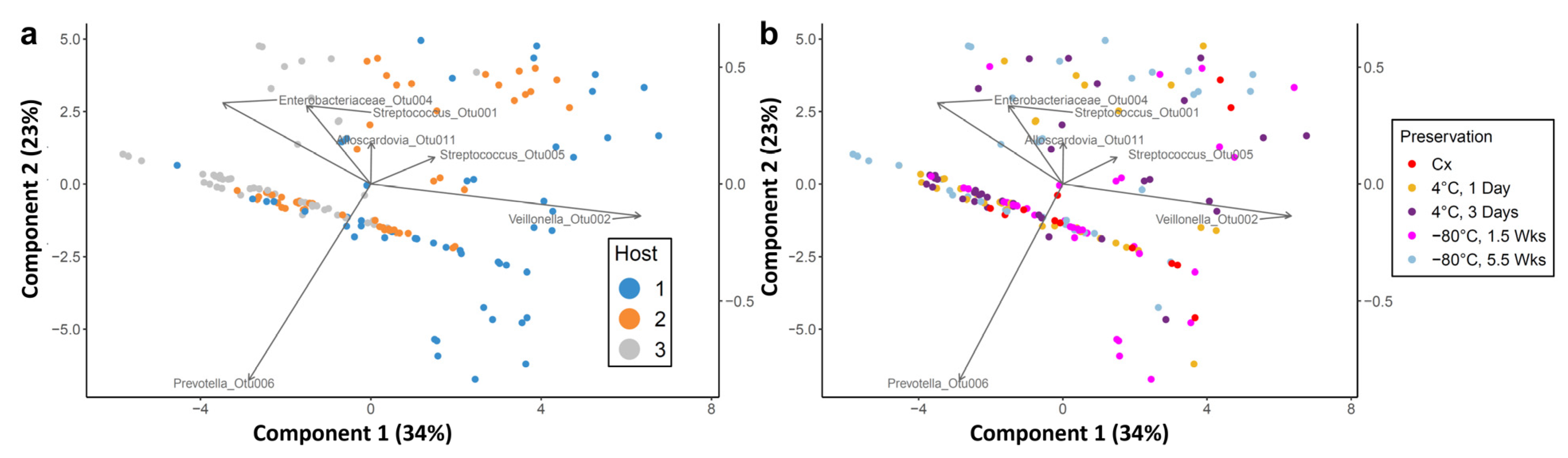
3.4. Propagation of Cold-Preserved Cultures Favors an “Attractor” Community Composition across Individual Samples
3.5. Composition of the “Attractor” Community
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dang, A.T.; Cotton, S.; Sankaran-Walters, S.; Li, C.-S.; Lee, C.-Y.M.; Dandekar, S.; Paster, B.J.; George, M.D. Evidence of an increased pathogenic footprint in the lingual microbiome of untreated HIV infected patients. BMC Microbiol. 2012, 12, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, S.M.; Haase Elaine, M.; Lesse Alan, J.; Gill Steven, R.; Scannapieco Frank, A. Genetic Relationships between Respiratory Pathogens Isolated from Dental Plaque and Bronchoalveolar Lavage Fluid from Patients in the Intensive Care Unit Undergoing Mechanical Ventilation. Clin. Infect. Dis. 2008, 47, 1562–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tada, A.; Hanada, N. Opportunistic respiratory pathogens in the oral cavity of the elderly: Oral microbial communities in the elderly population. FEMS Immunol. Med. Microbiol. 2010, 60, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Solh, A.A.; Pietrantoni, C.; Bhat, A.; Okada, M.; Zambon, J.; Aquilina, A.; Berbary, E. Colonization of Dental Plaques. Chest 2004, 126, 1575–1582. [Google Scholar] [CrossRef]
- Harper-Owen, R.; Dymock, D.; Booth, V.; Weightman, A.J.; Wade, W.G. Detection of Unculturable Bacteria in Periodontal Health and Disease by PCR. J. Clin. Microbiol. 1999, 37, 1469–1473. [Google Scholar] [CrossRef] [Green Version]
- Ximenez-Fyvie, L.A.; Almaguer-Flores, A.; Jacobo-Soto, V.; Lara-Cordoba, M.; Moreno-Borjas, J.-Y.; Alcantara-Maruri, E. Subgingival microbiota of periodontally untreated Mexican subjects with generalized aggressive periodontitis. J. Clin. Periodontol. 2006, 33, 869–877. [Google Scholar] [CrossRef]
- Senpuku, H.; Sogame, A.; Inoshita, E.; Tsuha, Y.; Miyazaki, H.; Hanada, N. Systemic Diseases in Association with Microbial Species in Oral Biofilm from Elderly Requiring Care. Gerontology 2003, 49, 301–309. [Google Scholar] [CrossRef]
- Lawrence, H.P. Salivary markers of systemic disease: Noninvasive diagnosis of disease and monitoring of general health. J. Can. Dent. Assoc. 2002, 68, 170–174. [Google Scholar]
- Mager, D.; Haffajee, A.; Devlin, P.; Norris, C.; Posner, M.; Goodson, J. The salivary microbiota as a diagnostic indicator of oral cancer: A descriptive, non-randomized study of cancer-free and oral squamous cell carcinoma subjects. J. Transl. Med. 2005, 3, 27. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Mocanu, V.; Cai, C.; Dang, J.; Slater, L.; Deehan, E.C.; Walter, J.; Madsen, K.L. Impact of Fecal Microbiota Transplantation on Obesity and Metabolic Syndrome—A Systematic Review. Nutrients 2019, 11, 2291. [Google Scholar] [CrossRef] [Green Version]
- Adler, E.; Tabaa, A.; Kassam, Z.; Zydek, M.; Terdiman, J.; El-Nachef, N. Capsule-Delivered Fecal Microbiota Transplant Is Safe and Well Tolerated in Patients with Ulcerative Colitis. Dig. Dis. Sci. 2019, 64, 2452–2454. [Google Scholar] [CrossRef] [PubMed]
- Cold, F.; Browne, P.D.; Günther, S.; Halkjaer, S.I.; Petersen, A.M.; Al-Gibouri, Z.; Hansen, L.H.; Christensen, A.H. Multidonor FMT capsules improve symptoms and decrease fecal calprotectin in ulcerative colitis patients while treated—An open-label pilot study. Scand. J. Gastroenterol. 2019, 54, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, L.d.F.; Borba, H.H.; Tonin, F.S.; Wiens, A.; Fernandez-Llimos, F.; Pontarolo, R. Fecal microbiota transplantation in inflammatory bowel disease patients: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0238910. [Google Scholar] [CrossRef] [PubMed]
- Paramsothy, S.; Kamm, M.A.; Kaakoush, N.O.; Walsh, A.J.; van den Bogaerde, J.; Samuel, D.; Leong, R.W.L.; Connor, S.; Ng, W.; Paramsothy, R.; et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: A randomised placebo-controlled trial. Lancet 2017, 389, 1218–1228. [Google Scholar] [CrossRef]
- Tian, Y.; Zhou, Y.; Huang, S.; Li, J.; Zhao, K.; Li, X.; Wen, X.; Li, X.-A. Fecal microbiota transplantation for ulcerative colitis: A prospective clinical study. BMC Gastroenterol. 2019, 19, 116. [Google Scholar] [CrossRef] [Green Version]
- Nath, S.; Zilm, P.; Jamieson, L.; Kapellas, K.; Goswami, N.; Ketagoda, K.; Weyrich, L.S. Development and characterization of an oral microbiome transplant among Australians for the treatment of dental caries and periodontal disease: A study protocol. PLoS ONE 2021, 16, e0260433. [Google Scholar] [CrossRef] [PubMed]
- Hope, C.K.; Clements, D.; Wilson, M. Determining the spatial distribution of viable and nonviable bacteria in hydrated microcosm dental plaques by viability profiling. J. Appl. Microbiol. 2002, 93, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Dige, I.; Nilsson, H.; Kilian, M.; Nyvad, B. In situ identification of streptococci and other bacteria in initial dental biofilm by confocal laser scanning microscopy and fluorescence in situ hybridization. Eur. J. Oral Sci. 2007, 115, 459–467. [Google Scholar] [CrossRef]
- Guggenheim, B.; Guggenheim, M.; Gmür, R.; Giertsen, E.; Thurnheer, T. Application of the Zürich Biofilm Model to Problems of Cariology. Caries Res. 2004, 38, 212–222. [Google Scholar] [CrossRef] [Green Version]
- Pratten, J.; Andrews, C.S.; Craig, D.Q.M.; Wilson, M. Structural studies of microcosm dental plaques grown under different nutritional conditions. FEMS Microbiol. Lett. 2000, 189, 215–218. [Google Scholar] [CrossRef]
- Abrams, M.; Barton, D.; Vandaei, E.; Romero, D.; Caldwell, A.; Ouverney, C. Genomic characteristics of an environmental microbial community harboring a novel human uncultured TM7 bacterium associated with oral diseases. Open Access Sci. Rep. 2012, 1, 276. [Google Scholar]
- Al-Ahmad, A.; Wunder, A.; Auschill, T.M.; Follo, M.; Braun, G.; Hellwig, E.; Arweiler, N.B. The in vivo dynamics of Streptococcus spp., Actinomyces naeslundii, Fusobacterium nucleatum and Veillonella spp. in dental plaque biofilm as analysed by five-colour multiplex fluorescence in situ hybridization. J. Med. Microbiol. 2007, 56, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, N.I.; Palmer, R.J.; Cisar, J.O.; Kolenbrander, P.E. Characterization of a Streptococcus sp.-Veillonella sp. Community Micromanipulated from Dental Plaque. J. Bacteriol. 2008, 190, 8145–8154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Ku, C.Y.S.; Xu, J.; Saxena, D.; Caufield, P.W. Survey of Oral Microbial Diversity using PCR-based Denaturing Gradient Gel Electrophoresis. J. Dent. Res. 2005, 84, 559–564. [Google Scholar] [CrossRef]
- Sizova, M.V.; Hohmann, T.; Hazen, A.; Paster, B.J.; Halem, S.R.; Murphy, C.M.; Panikov, N.S.; Epstein, S.S. New Approaches for Isolation of Previously Uncultivated Oral Bacteria. Appl. Environ. Microbiol. 2012, 78, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Vartoukian, S.R.; Adamowska, A.; Lawlor, M.; Moazzez, R.; Dewhirst, F.E.; Wade, W.G. In Vitro Cultivation of ‘Unculturable’ Oral Bacteria, Facilitated by Community Culture and Media Supplementation with Siderophores. PLoS ONE 2016, 11, e0146926. [Google Scholar] [CrossRef]
- Claassen, S.; du Toit, E.; Kaba, M.; Moodley, C.; Zar, H.J.; Nicol, M.P. A comparison of the efficiency of five different commercial DNA extraction kits for extraction of DNA from faecal samples. J. Microbiol. Methods 2013, 94, 103–110. [Google Scholar] [CrossRef]
- Tremblay, J.; Singh, K.; Fern, A.; Kirton, E.S.; He, S.; Woyke, T.; Lee, J.; Chen, F.; Dangl, J.L.; Tringe, S.G. Primer and platform effects on 16S rRNA tag sequencing. Front. Microbiol. 2015, 6, 771. [Google Scholar] [CrossRef] [Green Version]
- Huse, S.M.; Dethlefsen, L.; Huber, J.A.; Welch, D.M.; Relman, D.A.; Sogin, M.L. Exploring Microbial Diversity and Taxonomy Using SSU rRNA Hypervariable Tag Sequencing. PLoS Genet. 2008, 4, e1000255. [Google Scholar] [CrossRef]
- Liu, Z.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Res. 2008, 36, e120. [Google Scholar] [CrossRef] [Green Version]
- Mizrahi-Man, O.; Davenport, E.R.; Gilad, Y. Taxonomic Classification of Bacterial 16S rRNA Genes Using Short Sequencing Reads: Evaluation of Effective Study Designs. PLoS ONE 2013, 8, e53608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nossa, C.W. Design of 16S rRNA gene primers for 454 pyrosequencing of the human foregut microbiome. World J. Gastroenterol. 2010, 16, 4135. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gevers, D.; Knight, R.; Petrosino, J.F.; Huang, K.; McGuire, A.L.; Birren, B.W.; Nelson, K.E.; White, O.; Methé, B.A.; Huttenhower, C. The Human Microbiome Project: A Community Resource for the Healthy Human Microbiome. PLoS Biol. 2012, 10, e1001377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The Human Microbiome Project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, M.W.; Stadler, P.; Wu, Q.L.; Pöckl, M. The filtration–acclimatization method for isolation of an important fraction of the not readily cultivable bacteria. J. Microbiol. Methods 2004, 57, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Hayati, F.; Okada, A.; Kitasako, Y.; Tagami, J.; Matin, K. An artificial biofilm induced secondary caries model for in vitro studies: Biofilm induced secondary caries model. Aust. Dent. J. 2011, 56, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; He, X.; Torralba, M.; Yooseph, S.; Nelson, K.E.; Lux, R.; McLean, J.S.; Yu, G.; Shi, W. Using DGGE profiling to develop a novel culture medium suitable for oral microbial communities: DGGE profiling for culture medium development. Mol. Oral Microbiol. 2010, 25, 357–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinniment, S.L.; Wimpenny, J.W.T.; Adams, D.; Marsh, P.D. Development of a steady-state oral microbial biof ilrn community using the constant-depth film ferrnenter. Microbiology 1996, 142, 631–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kommerein, N.; Stumpp, S.N.; Müsken, M.; Ehlert, N.; Winkel, A.; Häussler, S.; Behrens, P.; Buettner, F.F.R.; Stiesch, M. An oral multispecies biofilm model for high content screening applications. PLoS ONE 2017, 12, e0173973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loesche, W.J.; Hockett, R.N.; Syed, S.A. The predominant cultivable flora of tooth surface plaque removed from institutionalized subjects. Arch. Oral Biol. 1972, 17, 1311–1325. [Google Scholar] [CrossRef] [Green Version]
- Lamont, E.I.; Gadkari, A.; Kerns, K.A.; To, T.T.; Daubert, D.; Kotsakis, G.; Bor, B.; He, X.; McLean, J.S. Modified SHI medium supports growth of a disease-state subgingival polymicrobial community in vitro. Mol. Oral Microbiol. 2021, 36, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Arthur, R.A.; Waeiss, R.A.; Hara, A.T.; Lippert, F.; Eckert, G.J.; Zero, D.T. A Defined-Multispecies Microbial Model for Studying Enamel Caries Development. Caries Res. 2013, 47, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, B.H.; Wefel, J.S.; Miller, I. A Model for Producing Caries-like Lesions in Enamel and Dentin Using Oral Bacteria in vitro. J. Dent. Res. 1984, 63, 1186–1189. [Google Scholar] [CrossRef]
- Guggenheim, B.; Giertsen, E.; Schüpbach, P.; Shapiro, S. Validation of an in vitro Biofilm Model of Supragingival Plaque. J. Dent. Res. 2001, 80, 363–370. [Google Scholar] [CrossRef]
- Steiner-Oliveira, C.; Rodrigues, L.K.A.; Zanin, I.; Carvalho, C.; Kamiya, R.; Hara, A.; Nobre-Dos-Santos, M. An in vitro microbial model associated with sucrose to produce dentin caries lesions. Open Life Sci. 2011, 6, 414–421. [Google Scholar] [CrossRef]
- Van de Sande, F.H.; Azevedo, M.S.; Lund, R.G.; Huysmans, M.C.D.N.J.M.; Cenci, M.S. An in vitro biofilm model for enamel demineralization and antimicrobial dose-response studies. Biofouling 2011, 27, 1057–1063. [Google Scholar] [CrossRef]
- Ceri, H.; Olson, M.E.; Stremick, C.; Read, R.R.; Morck, D.; Buret, A. The Calgary Biofilm Device: New Technology for Rapid Determination of Antibiotic Susceptibilities of Bacterial Biofilms. J. Clin. Microbiol. 1999, 37, 1771–1776. [Google Scholar] [CrossRef] [Green Version]
- Edlund, A.; Yang, Y.; Hall, A.P.; Guo, L.; Lux, R.; He, X.; Nelson, K.E.; Nealson, K.H.; Yooseph, S.; Shi, W.; et al. An in vitrobiofilm model system maintaining a highly reproducible species and metabolic diversity approaching that of the human oral microbiome. Microbiome 2013, 1, 25. [Google Scholar] [CrossRef] [Green Version]
- Filoche, S.K.; Soma, K.J.; Sissons, C.H. Caries-related plaque microcosm biofilms developed in microplates. Oral Microbiol. Immunol. 2007, 22, 73–79. [Google Scholar] [CrossRef]
- Sissons, C.H.; Cutress, T.W.; Hoffman, M.P.; Wakefield, J.S.J. A Multi-station Dental Plaque Microcosm (Artificial Mouth) for the Study of Plaque Growth, Metabolism, pH, and Mineralization. J. Dent. Res. 1991, 70, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.A.; Landerkin, G.B.; Morse, P.M. Effects of Additives on the Survival of Lactic Streptococci in Frozen Storage. Appl. Microbiol. 1966, 14, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto-Shinohara, Y.; Sukenobe, J.; Imaizumi, T.; Nakahara, T. Survival curves for microbial species stored by freeze-drying. Cryobiology 2006, 52, 27–32. [Google Scholar] [CrossRef]
- Choo, J.M.; Leong, L.E.; Rogers, G.B. Sample storage conditions significantly influence faecal microbiome profiles. Sci. Rep. 2015, 5, 16350. [Google Scholar] [CrossRef] [Green Version]
- Song, S.J.; Amir, A.; Metcalf, J.L.; Amato, K.R.; Xu, Z.Z.; Humphrey, G.; Knight, R. Preservation Methods Differ in Fecal Microbiome Stability, Affecting Suitability for Field Studies. mSystems 2016, 1, e00021-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedjo, D.I.; Jonkers, D.M.A.E.; Savelkoul, P.H.; Masclee, A.A.; van Best, N.; Pierik, M.J.; Penders, J. The Effect of Sampling and Storage on the Fecal Microbiota Composition in Healthy and Diseased Subjects. PLoS ONE 2015, 10, e0126685. [Google Scholar] [CrossRef] [PubMed]
- Bircher, L.; Geirnaert, A.; Hammes, F.; Lacroix, C.; Schwab, C. Effect of cryopreservation and lyophilization on viability and growth of strict anaerobic human gut microbes. Microb. Biotechnol. 2018, 11, 721–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, A.G.; Sim, K.; Powell, E.; Cornwell, E.; Cramer, T.; McClure, Z.E.; Li, M.S.; Kroll, J.S. Latitude in sample handling and storage for infant faecal microbiota studies: The elephant in the room? Microbiome 2016, 4, 40. [Google Scholar] [CrossRef] [Green Version]
- Reygner, J.; Charrueau, C.; Delannoy, J.; Mayeur, C.; Robert, V.; Cuinat, C.; Meylheuc, T.; Mauras, A.; Augustin, J.; Nicolis, I.; et al. Freeze-dried fecal samples are biologically active after long-lasting storage and suited to fecal microbiota transplantation in a preclinical murine model of Clostridioides difficile infection. Gut Microbes 2020, 11, 1405–1422. [Google Scholar] [CrossRef]
- Tap, J.; Cools-Portier, S.; Pavan, S.; Druesne, A.; Öhman, L.; Törnblom, H.; Simren, M.; Derrien, M. Effects of the long-term storage of human fecal microbiota samples collected in RNAlater. Sci. Rep. 2019, 9, 601. [Google Scholar] [CrossRef] [Green Version]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the MiSeq Illumina Sequencing Platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Peña, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples: Primers for marine microbiome studies. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the Effects of PCR Amplification and Sequencing Artifacts on 16S rRNA-Based Studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [Green Version]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.-A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [Green Version]
- Woese, C.R.; Fox, G.E. Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Proc. Natl. Acad. Sci. USA 1977, 74, 5088–5090. [Google Scholar] [CrossRef]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Luo, T.; Srinivasan, U.; Ramadugu, K.; Shedden, K.A.; Neiswanger, K.; Trumble, E.; Li, J.J.; McNeil, D.W.; Crout, R.J.; Weyant, R.J.; et al. Effects of Specimen Collection Methodologies and Storage Conditions on the Short-Term Stability of Oral Microbiome Taxonomy. Appl. Environ. Microbiol. 2016, 82, 5519–5529. [Google Scholar] [CrossRef] [Green Version]
- Hoover, C.I.; Newbrun, E. Survival of Bacteria from Human Dental Plaque Under Various Transport Conditions. J. Clin. Microbiol. 1977, 6, 212–218. [Google Scholar] [CrossRef]
- Mashima, I.; Nakazawa, F. The influence of oral Veillonella species on biofilms formed by Streptococcus species. Anaerobe 2014, 28, 54–61. [Google Scholar] [CrossRef]
- Krasse, B. The proportional distribution of Streptococcus salivarius and othe streptococci in various parts of the mouth. Odontol. Rev. 1954, 5, 203–211. [Google Scholar]
- Barbour, A.; Philip, K.; Muniandy, S. Enhanced Production, Purification, Characterization and Mechanism of Action of Salivaricin 9 Lantibiotic Produced by Streptococcus salivarius NU10. PLoS ONE 2013, 8, e77751. [Google Scholar] [CrossRef]
- Burton, J.P.; Wescombe, P.A.; Macklaim, J.M.; Chai, M.H.C.; Macdonald, K.; Hale, J.D.F.; Tagg, J.; Reid, G.; Gloor, G.B.; Cadieux, P.A. Persistence of the Oral Probiotic Streptococcus salivarius M18 Is Dose Dependent and Megaplasmid Transfer Can Augment Their Bacteriocin Production and Adhesion Characteristics. PLoS ONE 2013, 8, e65991. [Google Scholar] [CrossRef] [Green Version]
- Tompkins, G.R.; Tagg, J.R. The Ecology of Bacteriocin-producing Strains of Streptococcus salivarius. Microb. Ecol. Health Dis. 1989, 2, 19–28. [Google Scholar]
- Periasamy, S.; Kolenbrander, P.E. Central Role of the Early Colonizer Veillonella sp. in Establishing Multispecies Biofilm Communities with Initial, Middle, and Late Colonizers of Enamel. J. Bacteriol. 2010, 192, 2965–2972. [Google Scholar] [CrossRef] [Green Version]
- Mori, H.; Maruyama, F.; Kato, H.; Toyoda, A.; Dozono, A.; Ohtsubo, Y.; Nagata, Y.; Fujiyama, A.; Tsuda, M.; Kurokawa, K. Design and Experimental Application of a Novel Non-Degenerate Universal Primer Set that Amplifies Prokaryotic 16S rRNA Genes with a Low Possibility to Amplify Eukaryotic rRNA Genes. DNA Res. 2014, 21, 217–227. [Google Scholar] [CrossRef]
- Huys, G.; Vancanneyt, M.; D’Haene, K.; Falsen, E.; Wauters, G.; Vandamme, P. Alloscardovia omnicolens gen. nov., sp. nov., from human clinical samples. Int. J. Syst. Evol. Microbiol. 2007, 57, 1442–1446. [Google Scholar] [CrossRef] [Green Version]
- Mahlen, S.D.; Clarridge, J.E. Site and Clinical Significance of Alloscardovia omnicolens and Bifidobacterium Species Isolated in the Clinical Laboratory. J. Clin. Microbiol. 2009, 47, 3289–3293. [Google Scholar] [CrossRef]
- Tett, A.; Pasolli, E.; Masetti, G.; Ercolini, D.; Segata, N. Prevotella diversity, niches and interactions with the human host. Nat. Rev. Microbiol. 2021, 19, 585–599. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Schloss, P.D.; Handelsman, J. Introducing DOTUR, a Computer Program for Defining Operational Taxonomic Units and Estimating Species Richness. Appl. Environ. Microbiol. 2005, 71, 1501–1506. [Google Scholar] [CrossRef] [Green Version]
- Greenblum, S.; Carr, R.; Borenstein, E. Extensive Strain-Level Copy-Number Variation across Human Gut Microbiome Species. Cell 2015, 160, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Bihan, M.; Methé, B.A. Analyses of the Stability and Core Taxonomic Memberships of the Human Microbiome. PLoS ONE 2013, 8, e63139. [Google Scholar]
- Zhou, Y.; Gao, H.; Mihindukulasuriya, K.A.; La Rosa, P.S.; Wylie, K.M.; Vishnivetskaya, T.; Podar, M.; Warner, B.; Tarr, P.I.; Nelson, D.E.; et al. Biogeography of the ecosystems of the healthy human body. Genome Biol. 2013, 14, R1. [Google Scholar] [CrossRef] [Green Version]
- Thingholm, L.B.; Rühlemann, M.C.; Koch, M.; Fuqua, B.; Laucke, G.; Boehm, R.; Bang, C.; Franzosa, E.A.; Hübenthal, M.; Rahnavard, G.; et al. Obese Individuals with and without Type 2 Diabetes Show Different Gut Microbial Functional Capacity and Composition. Cell Host Microbe 2019, 26, 252–264.e10. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Torgerson, W. Theory and Methods of Scaling; Wiley: New York, NY, USA, 1958. [Google Scholar]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Austral. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Borcard, D.; Legendre, P. All-scale spatial analysis of ecological data by means of principal coordinates of neighbour matrices. Ecol. Model. 2002, 153, 51–68. [Google Scholar] [CrossRef]
- Borcard, D.; Legendre, P.; Drapeau, P. Partialling out the Spatial Component of Ecological Variation. Ecology 1992, 73, 1045–1055. [Google Scholar] [CrossRef] [Green Version]
- Kruskal, J.B. Multidimensional scaling by optimizing goodness of fit to a nonmetric hypothesis. Psychometrika 1964, 29, 1–27. [Google Scholar] [CrossRef]
- Pearson, K. LIII. On lines and planes of closest fit to systems of points in space. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1901, 2, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Wold, S.; Esbensen, K.; Geladi, P. Principal component analysis. Chemometr. Intell. Lab. Syst. 1987, 2, 37–52. [Google Scholar] [CrossRef]
- Galloway-Peña, J.; Hanson, B. Tools for Analysis of the Microbiome. Dig. Dis. Sci. 2020, 65, 674–685. [Google Scholar] [CrossRef] [Green Version]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef] [Green Version]
- Popova, C.; Dosseva-Panova, V.; Panov, V. Microbiology of Periodontal Diseases. A Review. Biotechnol. Biotechnol. Equip. 2013, 27, 3754–3759. [Google Scholar] [CrossRef]
- Caufield, P.W.; Schön, C.N.; Saraithong, P.; Li, Y.; Argimón, S. Oral Lactobacilli and Dental Caries: A Model for Niche Adaptation in Humans. J. Dent. Res. 2015, 94 (Suppl. 9), 110S–118S. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, B.; Mobberley, J.; Shi, K.; Chen, I.A. Effects of Preservation and Propagation Methodology on Microcosms Derived from the Oral Microbiome. Microorganisms 2022, 10, 2146. https://doi.org/10.3390/microorganisms10112146
Zhou B, Mobberley J, Shi K, Chen IA. Effects of Preservation and Propagation Methodology on Microcosms Derived from the Oral Microbiome. Microorganisms. 2022; 10(11):2146. https://doi.org/10.3390/microorganisms10112146
Chicago/Turabian StyleZhou, Baoqing, Jen Mobberley, Kelly Shi, and Irene A. Chen. 2022. "Effects of Preservation and Propagation Methodology on Microcosms Derived from the Oral Microbiome" Microorganisms 10, no. 11: 2146. https://doi.org/10.3390/microorganisms10112146