
Next Generation Sequencing Approaches to Characterize the Respiratory Tract Virome

 ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Human Virome
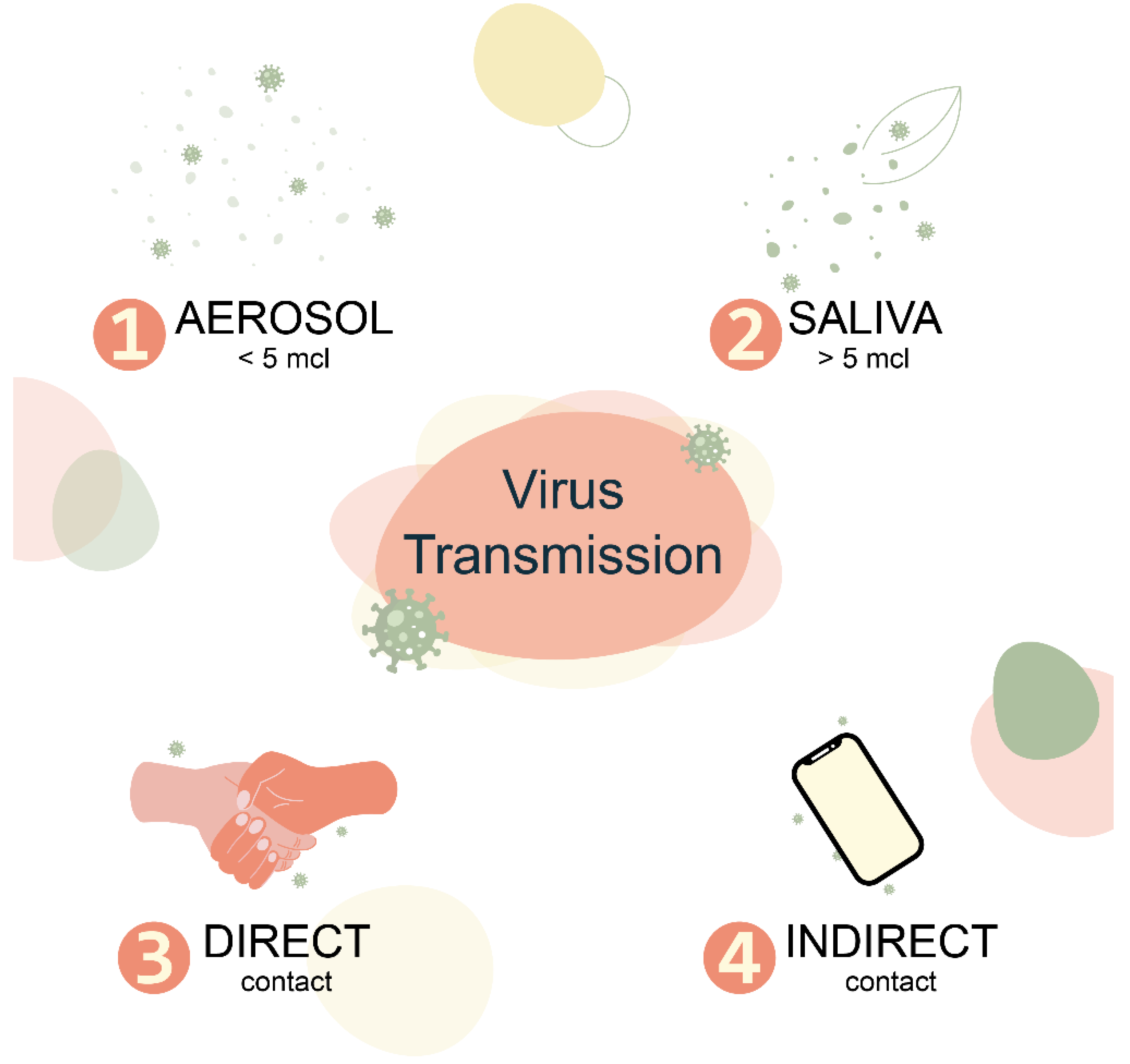
Human Respiratory Virome
3. ARVI as a Specific Case of Respiratory Virome Pathology
- Direct diagnostic methods for direct examination of biological material for the presence of a virus and/or viral antigen: electron microscopy (EM), enzyme immunoassay (ELISA), immunofluorescence reaction (RIF), radioimmunoassay (RIA), cytological methods;
- Direct molecular diagnostic methods for the presence of nucleic acids of viruses, based on DNA or RNA: PCR, RT-PCR, Real-Time PCR, sequencing, methods based on hybridization;
- Isolation and identification of the virus from clinical material in cell cultures;
- Serological diagnostics based on the determination of viral antibodies: complement fixation test (CFT), passive hemagglutination assay (PHA), indirect hemagglutination assay (IHA), and hemagglutination inhibition test (HI test).
4. NGS in the Human Respiratory Virome Study
4.1. NGS Sequencing Methods
4.1.1. Sample Collection
4.1.2. Virus Enrichment
4.1.3. Positive Selection Methods
4.1.4. Virus Enrichment Methods Based on Negative Exclusion
4.1.5. DNA/RNA Extraction Methods
4.1.6. Sequencing Quality Control
4.2. NGS Platforms for Virome Studies
4.3. Bioinformatics and NGS Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Weinbauer, M.G. Ecology of Prokaryotic Viruses. FEMS Microbiol. Rev. 2004, 28, 127–181. [Google Scholar] [CrossRef] [PubMed]
- Park, E.-J.; Kim, K.-H.; Abell, G.C.J.; Kim, M.-S.; Roh, S.W.; Bae, J.-W. Metagenomic Analysis of the Viral Communities in Fermented Foods. Appl. Environ. Microbiol. 2011, 77, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Taxonomic Information. Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 1 February 2022).
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Bracho, M.A.; Hillung, J.; García-González, N.; González-Candelas, F. High-Throughput Sequencing (HTS) for the Analysis of Viral Populations. Infect. Genet. Evol. 2020, 80, 104208. [Google Scholar] [CrossRef]
- Lecuit, M.; Eloit, M. The Human Virome: New Tools and Concepts. Trends Microbiol. 2013, 21, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Handelsman, J.; Rondon, M.R.; Brady, S.F.; Clardy, J.; Goodman, R.M. Molecular Biological Access to the Chemistry of Unknown Soil Microbes: A New Frontier for Natural Products. Chem. Biol. 1998, 5, R245–R249. [Google Scholar] [CrossRef]
- Dickins, B.; Nekrutenko, A. High-Resolution Mapping of Evolutionary Trajectories in a Phage. Genome Biol. Evol. 2009, 1, 294–307. [Google Scholar] [CrossRef]
- Wirbel, J.; Pyl, P.T.; Kartal, E.; Zych, K.; Kashani, A.; Milanese, A.; Fleck, J.S.; Voigt, A.Y.; Palleja, A.; Ponnudurai, R.; et al. Meta-Analysis of Fecal Metagenomes Reveals Global Microbial Signatures That Are Specific for Colorectal Cancer. Nat. Med. 2019, 25, 679–689. [Google Scholar] [CrossRef]
- Gorzelak, M.A.; Gill, S.K.; Tasnim, N.; Ahmadi-Vand, Z.; Jay, M.; Gibson, D.L. Methods for Improving Human Gut Microbiome Data by Reducing Variability through Sample Processing and Storage of Stool. PLoS ONE 2015, 10, e0134802. [Google Scholar] [CrossRef]
- Simner, P.J.; Miller, S.; Carroll, K.C. Understanding the Promises and Hurdles of Metagenomic Next-Generation Sequencing as a Diagnostic Tool for Infectious Diseases. Clin. Infect. Dis. 2018, 66, 778–788. [Google Scholar] [CrossRef]
- van der Helm, E.; Imamovic, L.; Hashim Ellabaan, M.M.; van Schaik, W.; Koza, A.; Sommer, M.O.A. Rapid Resistome Mapping Using Nanopore Sequencing. Nucleic Acids Res. 2017, 45, e61. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.K.; Ngo, T.T.; Tran, P.M.; Pham, T.T.T.; Vu, H.T.T.; Nguyen, N.T.H.; Thwaites, G.; Virtala, A.K.; Vapalahti, O.; Baker, S.; et al. Respiratory Viruses in Individuals with a High Frequency of Animal Exposure in Southern and Highland Vietnam. J. Med. Virol. 2020, 92, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Bai, G.-H.; Lin, S.-C.; Hsu, Y.-H.; Chen, S.-Y. The Human Virome: Viral Metagenomics, Relations with Human Diseases, and Therapeutic Applications. Viruses 2022, 14, 278. [Google Scholar] [CrossRef] [PubMed]
- Handley, S.A. The Virome: A Missing Component of Biological Interaction Networks in Health and Disease. Genome Med. 2016, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Virgin, H.W. The Virome in Mammalian Physiology and Disease. Cell 2014, 157, 142–150. [Google Scholar] [CrossRef]
- Moore, N.E.; Wang, J.; Hewitt, J.; Croucher, D.; Williamson, D.A.; Paine, S.; Yen, S.; Greening, G.E.; Hall, R.J. Metagenomic Analysis of Viruses in Feces from Unsolved Outbreaks of Gastroenteritis in Humans. J. Clin. Microbiol. 2015, 53, 15–21. [Google Scholar] [CrossRef]
- Deng, L.; Silins, R.; Castro-Mejía, J.L.; Kot, W.; Jessen, L.; Thorsen, J.; Shah, S.; Stokholm, J.; Bisgaard, H.; Moineau, S.; et al. A Protocol for Extraction of Infective Viromes Suitable for Metagenomics Sequencing from Low Volume Fecal Samples. Viruses 2019, 11, 667. [Google Scholar] [CrossRef]
- Mohammad, H.A.; Madi, N.M.; Al-Nakib, W. Analysis of Viral Diversity in Stool Samples from Infants and Children with Acute Gastroenteritis in Kuwait Using Metagenomics Approach. Virol. J. 2020, 17, 10. [Google Scholar] [CrossRef]
- Law, J.; Jovel, J.; Patterson, J.; Ford, G.; O’keefe, S.; Wang, W.; Meng, B.; Song, D.; Zhang, Y.; Tian, Z.; et al. Identification of Hepatotropic Viruses from Plasma Using Deep Sequencing: A Next Generation Diagnostic Tool. PLoS ONE 2013, 8, e60595. [Google Scholar] [CrossRef]
- Rascovan, N.; Duraisamy, R.; Desnues, C. Metagenomics and the Human Virome in Asymptomatic Individuals. Annu. Rev. Microbiol. 2016, 70, 125–141. [Google Scholar] [CrossRef]
- Miller, S.; Naccache, S.N.; Samayoa, E.; Messacar, K.; Arevalo, S.; Federman, S.; Stryke, D.; Pham, E.; Fung, B.; Bolosky, W.J.; et al. Laboratory Validation of a Clinical Metagenomic Sequencing Assay for Pathogen Detection in Cerebrospinal Fluid. Genome Res. 2019, 29, 831–842. [Google Scholar] [CrossRef]
- Wilson, M.R.; Sample, H.A.; Zorn, K.C.; Arevalo, S.; Yu, G.; Neuhaus, J.; Federman, S.; Stryke, D.; Briggs, B.; Langelier, C.; et al. Clinical Metagenomic Sequencing for Diagnosis of Meningitis and Encephalitis. N. Engl. J. Med. 2019, 380, 2327–2340. [Google Scholar] [CrossRef]
- Kohl, C.; Brinkmann, A.; Dabrowski, P.W.; Radonić, A.; Nitsche, A.; Kurth, A. Protocol for Metagenomic Virus Detection in Clinical Specimens1. Emerg. Infect. Dis. 2015, 21, 48–57. [Google Scholar] [CrossRef]
- Watson, M.; Schnettler, E.; Kohl, A. ViRome: An R Package for the Visualization and Analysis of Viral Small RNA Sequence Datasets. Bioinformatics 2013, 29, 1902–1903. [Google Scholar] [CrossRef]
- Lysholm, F.; Wetterbom, A.; Lindau, C.; Darban, H.; Bjerkner, A.; Fahlander, K.; Lindberg, A.M.; Persson, B.; Allander, T.; Andersson, B. Characterization of the Viral Microbiome in Patients with Severe Lower Respiratory Tract Infections, Using Metagenomic Sequencing. PLoS ONE 2012, 7, e30875. [Google Scholar] [CrossRef]
- Fischer, N.; Indenbirken, D.; Meyer, T.; Lütgehetmann, M.; Lellek, H.; Spohn, M.; Aepfelbacher, M.; Alawi, M.; Grundhoff, A. Evaluation of Unbiased Next-Generation Sequencing of RNA (RNA-Seq) as a Diagnostic Method in Influenza Virus-Positive Respiratory Samples. J. Clin. Microbiol. 2015, 53, 2238–2250. [Google Scholar] [CrossRef]
- Takeuchi, S.; Kawada, J.; Horiba, K.; Okuno, Y.; Okumura, T.; Suzuki, T.; Torii, Y.; Kawabe, S.; Wada, S.; Ikeyama, T.; et al. Metagenomic Analysis Using Next-Generation Sequencing of Pathogens in Bronchoalveolar Lavage Fluid from Pediatric Patients with Respiratory Failure. Sci. Rep. 2019, 9, 12909. [Google Scholar] [CrossRef]
- Li, Y.; Fu, X.; Ma, J.; Zhang, J.; Hu, Y.; Dong, W.; Wan, Z.; Li, Q.; Kuang, Y.-Q.; Lan, K.; et al. Altered Respiratory Virome and Serum Cytokine Profile Associated with Recurrent Respiratory Tract Infections in Children. Nat. Commun. 2019, 10, 2288. [Google Scholar] [CrossRef]
- van den Munckhof, E.H.A.; de Koning, M.N.C.; Quint, W.G.V.; van Doorn, L.-J.; Leverstein-van Hall, M.A. Evaluation of a Stepwise Approach Using Microbiota Analysis, Species-Specific QPCRs and Culture for the Diagnosis of Lower Respiratory Tract Infections. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 747–754. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-Specific Alterations in the Enteric Virome in Inflammatory Bowel Disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.Z.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A Highly Abundant Bacteriophage Discovered in the Unknown Sequences of Human Faecal Metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef]
- Abeles, S.R.; Ly, M.; Santiago-Rodriguez, T.M.; Pride, D.T. Effects of Long Term Antibiotic Therapy on Human Oral and Fecal Viromes. PLoS ONE 2015, 10, e0134941. [Google Scholar] [CrossRef]
- Wylie, K.M.; Mihindukulasuriya, K.A.; Zhou, Y.; Sodergren, E.; Storch, G.A.; Weinstock, G.M. Metagenomic Analysis of Double-Stranded DNA Viruses in Healthy Adults. BMC Biol. 2014, 12, 71. [Google Scholar] [CrossRef]
- Wylie, K.M. The Virome of the Human Respiratory Tract. Clin. Chest Med. 2017, 38, 11–19. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Nie, K.; Zhang, C.; Zhang, Y.; Wang, J.; Niu, P.; Ma, X. VIP: An Integrated Pipeline for Metagenomics of Virus Identification and Discovery. Sci. Rep. 2016, 6, 23774. [Google Scholar] [CrossRef]
- Graf, E.H.; Simmon, K.E.; Tardif, K.D.; Hymas, W.; Flygare, S.; Eilbeck, K.; Yandell, M.; Schlaberg, R. Unbiased Detection of Respiratory Viruses by Use of RNA Sequencing-Based Metagenomics: A Systematic Comparison to a Commercial PCR Panel. J. Clin. Microbiol. 2016, 54, 1000–1007. [Google Scholar] [CrossRef]
- Wylie, K.M.; Mihindukulasuriya, K.A.; Sodergren, E.; Weinstock, G.M.; Storch, G.A. Sequence Analysis of the Human Virome in Febrile and Afebrile Children. PLoS ONE 2012, 7, e27735. [Google Scholar] [CrossRef]
- Thorburn, F.; Bennett, S.; Modha, S.; Murdoch, D.; Gunson, R.; Murcia, P.R. The Use of next Generation Sequencing in the Diagnosis and Typing of Respiratory Infections. J. Clin. Virol. 2015, 69, 96–100. [Google Scholar] [CrossRef]
- Jain, S.; Self, W.H.; Wunderink, R.G.; Fakhran, S.; Balk, R.; Bramley, A.M.; Reed, C.; Grijalva, C.G.; Anderson, E.J.; Courtney, D.M.; et al. Community-Acquired Pneumonia Requiring Hospitalization among U.S. Adults. N. Engl. J. Med. 2015, 373, 415–427. [Google Scholar] [CrossRef]
- Flight, W.G.; Bright-Thomas, R.J.; Tilston, P.; Mutton, K.J.; Guiver, M.; Morris, J.; Webb, A.K.; Jones, A.M. Incidence and Clinical Impact of Respiratory Viruses in Adults with Cystic Fibrosis. Thorax 2014, 69, 247–253. [Google Scholar] [CrossRef]
- Goffard, A.; Lambert, V.; Salleron, J.; Herwegh, S.; Engelmann, I.; Pinel, C.; Pin, I.; Perrez, T.; Prévotat, A.; Dewilde, A.; et al. Virus and Cystic Fibrosis: Rhinoviruses Are Associated with Exacerbations in Adult Patients. J. Clin. Virol. 2014, 60, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Hedegaard, J.; Thorsen, K.; Lund, M.K.; Hein, A.-M.K.; Hamilton-Dutoit, S.J.; Vang, S.; Nordentoft, I.; Birkenkamp-Demtröder, K.; Kruhøffer, M.; Hager, H.; et al. Next-Generation Sequencing of RNA and DNA Isolated from Paired Fresh-Frozen and Formalin-Fixed Paraffin-Embedded Samples of Human Cancer and Normal Tissue. PLoS ONE 2014, 9, e98187. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Deveson, I.W.; Pang, C.N.I.; Yeang, M.; Naing, Z.; Adikari, T.; Hammond, J.M.; Stevanovski, I.; Beukers, A.G.; Verich, A.; et al. Respiratory Viral Co-Infections among SARS-CoV-2 Cases Confirmed by Virome Capture Sequencing. Sci. Rep. 2021, 11, 3934. [Google Scholar] [CrossRef] [PubMed]
- Forum of International Respiratory Societies. In The Global Impact of Respiratory Disease; European Respiratory Society: Sheffield, UK, 2017; ISBN 9781849840880.
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and Regional Mortality from 235 Causes of Death for 20 Age Groups in 1990 and 2010: A Systematic Analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Nair, H.; Simões, E.A.; Rudan, I.; Gessner, B.D.; Azziz-Baumgartner, E.; Zhang, J.S.F.; Feikin, D.R.; Mackenzie, G.A.; Moiïsi, J.C.; Roca, A.; et al. Global and Regional Burden of Hospital Admissions for Severe Acute Lower Respiratory Infections in Young Children in 2010: A Systematic Analysis. Lancet 2013, 381, 1380–1390. [Google Scholar] [CrossRef]
- Bates, M.; Mudenda, V.; Mwaba, P.; Zumla, A. Deaths Due to Respiratory Tract Infections in Africa. Curr. Opin. Pulm. Med. 2013, 19, 229–237. [Google Scholar] [CrossRef]
- van Doorn, H.R.; Yu, H. Viral Respiratory Infections. Hunter’s Trop. Med. Emerg. Infect. Dis. 2013, 269–274. [Google Scholar] [CrossRef]
- Leung, N.H.L. Transmissibility and Transmission of Respiratory Viruses. Nat. Rev. Microbiol. 2021, 19, 528–545. [Google Scholar] [CrossRef]
- Up to 650,000 People Die of Respiratory Diseases Linked to Seasonal Flu Each Year. Available online: https://www.who.int/news/item/13-12-2017-up-to-650-000-people-die-of-respiratory-diseases-linked-to-seasonal-flu-each-year (accessed on 2 February 2022).
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 2 February 2022).
- Kazakhstan: Coronavirus Pandemic Country Profile. Available online: https://ourworldindata.org/coronavirus/country/kazakhstan#how-many-tests-are-performed-each-day (accessed on 2 February 2022).
- World Health Organization. Zoonoses—Fact Sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/zoonoses (accessed on 2 February 2022).
- World Health Organization. Sars (Severe Acute Respiratory Syndrome). Available online: https://www.who.int/health-topics/severe-acute-respiratory-syndrome#tab=tab_1 (accessed on 2 February 2022).
- Heikkinen, T.; Järvinen, A. The Common Cold. Lancet 2003, 361, 51–59. [Google Scholar] [CrossRef]
- Brankston, G.; Gitterman, L.; Hirji, Z.; Lemieux, C.; Gardam, M. Transmission of Influenza A in Human Beings. Lancet Infect. Dis. 2007, 7, 257–265. [Google Scholar] [CrossRef]
- Brooks, W.A.; Goswami, D.; Rahman, M.; Nahar, K.; Fry, A.M.; Balish, A.; Iftekharuddin, N.; Azim, T.; Xu, X.; Klimov, A.; et al. Influenza Is a Major Contributor to Childhood Pneumonia in a Tropical Developing Country. Pediatr. Infect. Dis. J. 2010, 29, 216–221. [Google Scholar] [CrossRef]
- Janssens, J.P.; Krause, K.H. Pneumonia in the Very Old. Lancet Infect. Dis. 2004, 4, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Levine, O.S.; O’Brien, K.L.; Deloria-Knoll, M.; Murdoch, D.R.; Feikin, D.R.; Deluca, A.N.; Driscoll, A.J.; Baggett, H.C.; Brooks, W.A.; Howie, S.R.C.; et al. The Pneumonia Etiology Research for Child Health Project: A 21st Century Childhood Pneumonia Etiology Study. Clin. Infect. Dis. 2012, 54, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Rudan, I.; Boschi-Pinto, C.; Biloglav, Z.; Mulholland, K.; Campbell, H. Epidemiology and Etiology of Childhood Pneumonia. Bull. World Health Organ. 2008, 86, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Lin, S.; Zhang, H.; Liang, L.; Shen, S. Methods of Respiratory Virus Detection: Advances towards Point-of-Care for Early Intervention. Micromachines 2021, 12, 697. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, S.; Peter, J.; Xu, Z.; Bordoloi, D.; Ho, M.; Kalyanaraman, V.S.; Srinivasan, A.; Muthumani, K. Landscape of Humoral Immune Responses against SARS-CoV-2 in Patients with COVID-19 Disease and the Value of Antibody Testing. Heliyon 2021, 7, e06836. [Google Scholar] [CrossRef]
- Long, Q.-X.; Liu, B.-Z.; Deng, H.-J.; Wu, G.-C.; Deng, K.; Chen, Y.-K.; Liao, P.; Qiu, J.-F.; Lin, Y.; Cai, X.-F.; et al. Antibody Responses to SARS-CoV-2 in Patients with COVID-19. Nat. Med. 2020, 26, 845–848. [Google Scholar] [CrossRef]
- Gorse, G.J.; Donovan, M.M.; Patel, G.B. Antibodies to Coronaviruses Are Higher in Older Compared with Younger Adults and Binding Antibodies Are More Sensitive than Neutralizing Antibodies in Identifying Coronavirus-associated Illnesses. J. Med. Virol. 2020, 92, 512–517. [Google Scholar] [CrossRef]
- Quiñones-Mateu, M.E.; Avila, S.; Reyes-Teran, G.; Martinez, M.A. Deep Sequencing: Becoming a Critical Tool in Clinical Virology. J. Clin. Virol. 2014, 61, 9–19. [Google Scholar] [CrossRef]
- Grard, G.; Fair, J.N.; Lee, D.; Slikas, E.; Steffen, I.; Muyembe, J.-J.; Sittler, T.; Veeraraghavan, N.; Ruby, J.G.; Wang, C.; et al. A Novel Rhabdovirus Associated with Acute Hemorrhagic Fever in Central Africa. PLoS Pathog. 2012, 8, e1002924. [Google Scholar] [CrossRef]
- Van Tan, L.; van Doorn, H.R.; Nghia, H.D.T.; Chau, T.T.H.; Tu, L.T.P.; de Vries, M.; Canuti, M.; Deijs, M.; Jebbink, M.F.; Baker, S.; et al. Identification of a New Cyclovirus in Cerebrospinal Fluid of Patients with Acute Central Nervous System Infections. mBio 2013, 4, e00231-13. [Google Scholar] [CrossRef] [PubMed]
- van Boheemen, S.; van Rijn, A.L.; Pappas, N.; Carbo, E.C.; Vorderman, R.H.P.; Sidorov, I.; van’t Hof, P.J.; Mei, H.; Claas, E.C.J.; Kroes, A.C.M.; et al. Retrospective Validation of a Metagenomic Sequencing Protocol for Combined Detection of RNA and DNA Viruses Using Respiratory Samples from Pediatric Patients. J. Mol. Diagn. 2020, 22, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Mongkolrattanothai, K.; Naccache, S.N.; Bender, J.M.; Samayoa, E.; Pham, E.; Yu, G.; Dien Bard, J.; Miller, S.; Aldrovandi, G.; Chiu, C.Y. Neurobrucellosis: Unexpected Answer From Metagenomic Next-Generation Sequencing. J. Pediatr. Infect. Dis. Soc. 2017, 6, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Takayama, I.; Nguyen, B.G.; Dao, C.X.; Pham, T.T.; Dang, T.Q.; Truong, P.T.; Van Do, T.; Pham, T.T.P.; Fujisaki, S.; Odagiri, T.; et al. Next-Generation Sequencing Analysis of the Within-Host Genetic Diversity of Influenza A(H1N1)Pdm09 Viruses in the Upper and Lower Respiratory Tracts of Patients with Severe Influenza. mSphere 2021, 6, e01043-20. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Cai, Q.; Miao, Q.; Song, Z.; Fang, Y.; Hu, B. High-Throughput Metagenomics for Identification of Pathogens in the Clinical Settings. Small Methods 2021, 5, 2000792. [Google Scholar] [CrossRef]
- Sanjuán, R. Viral Mutation Rates. In Virus Evolution: Current Research and Future Directions; Caister Academic Press: Norfolk, UK, 2016; pp. 1–28. [Google Scholar]
- Parker, J.; Chen, J. Application of next Generation Sequencing for the Detection of Human Viral Pathogens in Clinical Specimens. J. Clin. Virol. 2017, 86, 20–26. [Google Scholar] [CrossRef]
- Zou, X.; Tang, G.; Zhao, X.; Huang, Y.; Chen, T.; Lei, M.; Chen, W.; Yang, L.; Zhu, W.; Zhuang, L.; et al. Simultaneous Virus Identification and Characterization of Severe Unexplained Pneumonia Cases Using a Metagenomics Sequencing Technique. Sci. China Life Sci. 2017, 60, 279–286. [Google Scholar] [CrossRef][Green Version]
- Loeffelholz, M.J.; Tang, Y.-W. Laboratory Diagnosis of Emerging Human Coronavirus Infections—The State of the Art. Emerg. Microbes Infect. 2020, 9, 747–756. [Google Scholar] [CrossRef]
- Abu-Diab, A.; Azzeh, M.; Ghneim, R.; Ghneim, R.; Zoughbi, M.; Turkuman, S.; Rishmawi, N.; Issa, A.-E.-R.; Siriani, I.; Dauodi, R.; et al. Comparison between Pernasal Flocked Swabs and Nasopharyngeal Aspirates for Detection of Common Respiratory Viruses in Samples from Children. J. Clin. Microbiol. 2008, 46, 2414–2417. [Google Scholar] [CrossRef]
- Agoritsas, K.; Mack, K.; Bonsu, B.K.; Goodman, D.; Salamon, D.; Marcon, M.J. Evaluation of the Quidel QuickVue Test for Detection of Influenza A and B Viruses in the Pediatric Emergency Medicine Setting by Use of Three Specimen Collection Methods. J. Clin. Microbiol. 2006, 44, 2638–2641. [Google Scholar] [CrossRef][Green Version]
- Gruteke, P.; Glas, A.S.; Dierdorp, M.; Vreede, W.B.; Pilon, J.-W.; Bruisten, S.M. Practical Implementation of a Multiplex PCR for Acute Respiratory Tract Infections in Children. J. Clin. Microbiol. 2004, 42, 5596–5603. [Google Scholar] [CrossRef][Green Version]
- Sung, R.Y.T.; Chan, P.K.S.; Choi, K.C.; Yeung, A.C.M.; Li, A.M.; Tang, J.W.; Ip, M.; Tsen, T.; Nelson, E.A.S. Comparative Study of Nasopharyngeal Aspirate and Nasal Swab Specimens for Diagnosis of Acute Viral Respiratory Infection. J. Clin. Microbiol. 2008, 46, 3073–3076. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.E.; Beekmann, S.E.; Polgreen, P.; Poser, S.; St. Pierre, J.; Santibañez, S.; Gerber, S.I.; Kim, L. Survey of Diagnostic Testing for Respiratory Syncytial Virus (RSV) in Adults: Infectious Disease Physician Practices and Implications for Burden Estimates. Diagn. Microbiol. Infect. Dis. 2018, 92, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-J.; Arnold, J.C.; Fairchok, M.P.; Danaher, P.J.; McDonough, E.A.; Blair, P.J.; Garcia, J.; Halsey, E.S.; Schofield, C.; Ottolini, M.; et al. Epidemiologic, Clinical, and Virologic Characteristics of Human Rhinovirus Infection among Otherwise Healthy Children and Adults. J. Clin. Virol. 2015, 64, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Bochkov, Y.A.; Gern, J.E. Clinical and Molecular Features of Human Rhinovirus C. Microbes Infect. 2012, 14, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Ferravante, C.; Sanna, G.; Melone, V.; Fromentier, A.; Rocco, T.; D’Agostino, Y.; Lamberti, J.; Alexandrova, E.; Pecoraro, G.; Pagliano, P.; et al. Nasopharyngeal Virome Analysis of COVID-19 Patients during Three Different Waves in Campania Region of Italy. J. Med. Virol. 2022, 94, 2275–2283. [Google Scholar] [CrossRef]
- Terlizzi, M.E.; Massimiliano, B.; Francesca, S.; Sinesi, F.; Rosangela, V.; Stefano, G.; Costa, C.; Rossana, C. Quantitative RT Real Time PCR and Indirect Immunofluorescence for the Detection of Human Parainfluenza Virus 1, 2, 3. J. Virol. Methods 2009, 160, 172–177. [Google Scholar] [CrossRef]
- Boivin, G.; Osterhaus, A.D.; Gaudreau, A.; Jackson, H.C.; Groen, J.; Ward, P. Roleof Picornaviruses in Flu-Like Illnesses of Adults Enrolled in AnOseltamivir Treatment Study Who Had No Evidence of Influenza VirusInfection. J. Clin. Microbiol. 2002, 40, 330–334. [Google Scholar] [CrossRef][Green Version]
- Weinberg, G.A.; Erdman, D.D.; Edwards, K.M.; Hall, C.B.; Walker, F.J.; Griffin, M.R.; Schwartz, B. Superiority of Reverse-Transcription Polymerase Chain Reaction to Conventional Viral Culture in the Diagnosis of Acute Respiratory Tract Infections in Children. J. Infect. Dis. 2004, 189, 706–710. [Google Scholar] [CrossRef][Green Version]
- Dwyer, D.E.; McPhie, K.A.; Ratnamohan, V.M.; Pitman, C.N. Challenges for the Laboratory before and during an Influenza Pandemic. N. S. W. Public Health Bull. 2006, 17, 142–145. [Google Scholar] [CrossRef]
- Covalciuc, K.A.; Webb, K.H.; Carlson, C.A. Comparison of Four Clinical Specimen Types for Detection of Influenza A and B Viruses by Optical Immunoassay (FLU OIA Test) and Cell Culture Methods. J. Clin. Microbiol. 1999, 37, 3971–3974. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, G.; Embree, J.; McNicol, P.; Law, B.; Hammond, G.W. Comparison of Nasopharyngeal Aspirate and Nasopharyngeal Swab Specimens for Respiratory Syncytial Virus Diagnosis by Cell Culture, Indirect Immunofluorescence Assay, and Enzyme-Linked Immunosorbent Assay. J. Clin. Microbiol. 1987, 25, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Yahia, S.; Kandeel, A.Y.; Hammad, E.; El-Gilany, A.-H. Human Metapneumovirus (HMPV) in Acute Respiratory Infection: A Clinic-Based Study in Egypt. Indian J. Pediatr. 2012, 79, 1323–1327. [Google Scholar] [CrossRef]
- van Rijn, A.L.; van Boheemen, S.; Sidorov, I.; Carbo, E.C.; Pappas, N.; Mei, H.; Feltkamp, M.; Aanerud, M.; Bakke, P.; Claas, E.C.J.; et al. The Respiratory Virome and Exacerbations in Patients with Chronic Obstructive Pulmonary Disease. PLoS ONE 2019, 14, e0223952. [Google Scholar] [CrossRef] [PubMed]
- Kustin, T.; Ling, G.; Sharabi, S.; Ram, D.; Friedman, N.; Zuckerman, N.; Bucris, E.D.; Glatman-Freedman, A.; Stern, A.; Mandelboim, M. A Method to Identify Respiratory Virus Infections in Clinical Samples Using Next-Generation Sequencing. Sci. Rep. 2019, 9, 2606. [Google Scholar] [CrossRef] [PubMed]
- Bharucha, T.; Oeser, C.; Balloux, F.; Brown, J.R.; Carbo, E.C.; Charlett, A.; Chiu, C.Y.; Claas, E.C.J.; de Goffau, M.C.; de Vries, J.J.C.; et al. STROBE-Metagenomics: A STROBE Extension Statement to Guide the Reporting of Metagenomics Studies. Lancet Infect. Dis. 2020, 20, e251–e260. [Google Scholar] [CrossRef]
- van Elden, L.J.R.; van Loon, A.M.; van Alphen, F.; Hendriksen, K.A.W.; Hoepelman, A.I.M.; van Kraaij, M.G.J.; Oosterheert, J.; Schipper, P.; Schuurman, R.; Nijhuis, M. Frequent Detection of Human Coronaviruses in Clinical Specimens from Patients with Respiratory Tract Infection by Use of a Novel Real-Time Reverse-Transcriptase Polymerase Chain Reaction. J. Infect. Dis. 2004, 189, 652–657. [Google Scholar] [CrossRef]
- Xu, B.; Liu, L.; Huang, X.; Ma, H.; Zhang, Y.; Du, Y.; Wang, P.; Tang, X.; Wang, H.; Kang, K.; et al. Metagenomic Analysis of Fever, Thrombocytopenia and Leukopenia Syndrome (FTLS) in Henan Province, China: Discovery of a New Bunyavirus. PLoS Pathog. 2011, 7, e1002369. [Google Scholar] [CrossRef]
- Li, C.-X.; Li, W.; Zhou, J.; Zhang, B.; Feng, Y.; Xu, C.-P.; Lu, Y.-Y.; Holmes, E.C.; Shi, M. High Resolution Metagenomic Characterization of Complex Infectomes in Paediatric Acute Respiratory Infection. Sci. Rep. 2020, 10, 3963. [Google Scholar] [CrossRef]
- Seelenfreund, E.; Robinson, W.A.; Amato, C.M.; Tan, A.-C.; Kim, J.; Robinson, S.E. Long Term Storage of Dry versus Frozen RNA for Next Generation Molecular Studies. PLoS ONE 2014, 9, e111827. [Google Scholar] [CrossRef]
- Vibin, J.; Chamings, A.; Collier, F.; Klaassen, M.; Nelson, T.M.; Alexandersen, S. Metagenomics Detection and Characterisation of Viruses in Faecal Samples from Australian Wild Birds. Sci. Rep. 2018, 8, 8686. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Murthy, S.; Kapoor, A. Evolution of Selective-Sequencing Approaches for Virus Discovery and Virome Analysis. Virus Res. 2017, 239, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Conceição-Neto, N.; Zeller, M.; Lefrère, H.; De Bruyn, P.; Beller, L.; Deboutte, W.; Yinda, C.K.; Lavigne, R.; Maes, P.; Van Ranst, M.; et al. Modular Approach to Customise Sample Preparation Procedures for Viral Metagenomics: A Reproducible Protocol for Virome Analysis. Sci. Rep. 2015, 5, 16532. [Google Scholar] [CrossRef] [PubMed]
- Allander, T.; Emerson, S.U.; Engle, R.E.; Purcell, R.H.; Bukh, J. A Virus Discovery Method Incorporating DNase Treatment and Its Application to the Identification of Two Bovine Parvovirus Species. Proc. Natl. Acad. Sci. USA 2001, 98, 11609–11614. [Google Scholar] [CrossRef] [PubMed]
- Rosseel, T.; Ozhelvaci, O.; Freimanis, G.; Van Borm, S. Evaluation of Convenient Pretreatment Protocols for RNA Virus Metagenomics in Serum and Tissue Samples. J. Virol. Methods 2015, 222, 72–80. [Google Scholar] [CrossRef]
- Fitzpatrick, A.H.; Rupnik, A.; O’Shea, H.; Crispie, F.; Keaveney, S.; Cotter, P. High Throughput Sequencing for the Detection and Characterization of RNA Viruses. Front. Microbiol. 2021, 12, 621719. [Google Scholar] [CrossRef]
- Davis, J.J.; Long, S.W.; Christensen, P.A.; Olsen, R.J.; Olson, R.; Shukla, M.; Subedi, S.; Stevens, R.; Musser, J.M. Analysis of the ARTIC Version 3 and Version 4 SARS-CoV-2 Primers and Their Impact on the Detection of the G142D Amino Acid Substitution in the Spike Protein. Microbiol. Spectr. 2021, 9, e01803-21. [Google Scholar] [CrossRef]
- Lambisia, A.W.; Mohammed, K.S.; Makori, T.O.; Ndwiga, L.; Mburu, M.W.; Morobe, J.M.; Moraa, E.O.; Musyoki, J.; Murunga, N.; Mwangi, J.N.; et al. Optimization of the SARS-CoV-2 ARTIC Network V4 Primers and Whole Genome Sequencing Protocol. Front. Med. 2022, 9, 836728. [Google Scholar] [CrossRef]
- Gardner, S.N.; Jaing, C.J.; McLoughlin, K.S.; Slezak, T.R. A Microbial Detection Array (MDA) for Viral and Bacterial Detection. BMC Genom. 2010, 11, 668. [Google Scholar] [CrossRef]
- Briese, T.; Kapoor, A.; Mishra, N.; Jain, K.; Kumar, A.; Jabado, O.J.; Lipkin, W.I. Virome Capture Sequencing Enables Sensitive Viral Diagnosis and Comprehensive Virome Analysis. mBio 2015, 6, e01491-15. [Google Scholar] [CrossRef]
- Wylie, T.N.; Wylie, K.M.; Herter, B.N.; Storch, G.A. Enhanced Virome Sequencing Using Targeted Sequence Capture. Genome Res. 2015, 25, 1910–1920. [Google Scholar] [CrossRef] [PubMed]
- Cholleti, H.; Hayer, J.; Fafetine, J.; Berg, M.; Blomström, A.-L. Genetic Characterization of a Novel Picorna-like Virus in Culex Spp. Mosquitoes from Mozambique. Virol. J. 2018, 15, 71. [Google Scholar] [CrossRef]
- Zhao, L.; Niu, Y.; Lu, T.; Yin, H.; Zhang, Y.; Xu, L.; Wang, Y.; Chen, H. Metagenomic Analysis of the Jinding Duck Fecal Virome. Curr. Microbiol. 2018, 75, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Strubbia, S.; Schaeffer, J.; Oude Munnink, B.B.; Besnard, A.; Phan, M.V.T.; Nieuwenhuijse, D.F.; de Graaf, M.; Schapendonk, C.M.E.; Wacrenier, C.; Cotten, M.; et al. Metavirome Sequencing to Evaluate Norovirus Diversity in Sewage and Related Bioaccumulated Oysters. Front. Microbiol. 2019, 10, 2394. [Google Scholar] [CrossRef] [PubMed]
- Sabatier, M.; Bal, A.; Destras, G.; Regue, H.; Quéromès, G.; Cheynet, V.; Lina, B.; Bardel, C.; Brengel-Pesce, K.; Navratil, V.; et al. Comparison of Nucleic Acid Extraction Methods for a Viral Metagenomics Analysis of Respiratory Viruses. Microorganisms 2020, 8, 1539. [Google Scholar] [CrossRef]
- Lewandowska, D.W.; Zagordi, O.; Geissberger, F.-D.; Kufner, V.; Schmutz, S.; Böni, J.; Metzner, K.J.; Trkola, A.; Huber, M. Optimization and Validation of Sample Preparation for Metagenomic Sequencing of Viruses in Clinical Samples. Microbiome 2017, 5, 94. [Google Scholar] [CrossRef]
- Klenner, J.; Kohl, C.; Dabrowski, P.W.; Nitsche, A. Comparing Viral Metagenomic Extraction Methods. Curr. Issues Mol. Biol. 2017, 24, 59–70. [Google Scholar] [CrossRef]
- Zhang, D.; Lou, X.; Yan, H.; Pan, J.; Mao, H.; Tang, H.; Shu, Y.; Zhao, Y.; Liu, L.; Li, J.; et al. Metagenomic Analysis of Viral Nucleic Acid Extraction Methods in Respiratory Clinical Samples. BMC Genom. 2018, 19, 773. [Google Scholar] [CrossRef]
- Thoendel, M.; Jeraldo, P.; Greenwood-Quaintance, K.E.; Yao, J.; Chia, N.; Hanssen, A.D.; Abdel, M.P.; Patel, R. Impact of Contaminating DNA in Whole-Genome Amplification Kits Used for Metagenomic Shotgun Sequencing for Infection Diagnosis. J. Clin. Microbiol. 2017, 55, 1789–1801. [Google Scholar] [CrossRef]
- Drengenes, C.; Wiker, H.G.; Kalananthan, T.; Nordeide, E.; Eagan, T.M.L.; Nielsen, R. Laboratory Contamination in Airway Microbiome Studies. BMC Microbiol. 2019, 19, 187. [Google Scholar] [CrossRef]
- Stinson, L.F.; Keelan, J.A.; Payne, M.S. Identification and Removal of Contaminating Microbial DNA from PCR Reagents: Impact on Low-biomass Microbiome Analyses. Lett. Appl. Microbiol. 2019, 68, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.R.; Uyaguari-Diaz, M.; McCabe, M.N.; Montoya, V.; Gardy, J.L.; Parker, S.; Steiner, T.; Hsiao, W.; Nesbitt, M.J.; Tang, P.; et al. Metagenomic Investigation of Plasma in Individuals with ME/CFS Highlights the Importance of Technical Controls to Elucidate Contamination and Batch Effects. PLoS ONE 2016, 11, e0165691. [Google Scholar] [CrossRef] [PubMed]
- Gargis, A.S.; Kalman, L.; Lubin, I.M. Assuring the Quality of Next-Generation Sequencing in Clinical Microbiology and Public Health Laboratories. J. Clin. Microbiol. 2016, 54, 2857–2865. [Google Scholar] [CrossRef] [PubMed]
- Asplund, M.; Kjartansdóttir, K.R.; Mollerup, S.; Vinner, L.; Fridholm, H.; Herrera, J.A.R.; Friis-Nielsen, J.; Hansen, T.A.; Jensen, R.H.; Nielsen, I.B.; et al. Contaminating Viral Sequences in High-Throughput Sequencing Viromics: A Linkage Study of 700 Sequencing Libraries. Clin. Microbiol. Infect. 2019, 25, 1277–1285. [Google Scholar] [CrossRef]
- Glassing, A.; Dowd, S.E.; Galandiuk, S.; Davis, B.; Chiodini, R.J. Inherent Bacterial DNA Contamination of Extraction and Sequencing Reagents May Affect Interpretation of Microbiota in Low Bacterial Biomass Samples. Gut Pathog. 2016, 8, 24. [Google Scholar] [CrossRef]
- Bal, A.; Pichon, M.; Picard, C.; Casalegno, J.S.; Valette, M.; Schuffenecker, I.; Billard, L.; Vallet, S.; Vilchez, G.; Cheynet, V.; et al. Quality Control Implementation for Universal Characterization of DNA and RNA Viruses in Clinical Respiratory Samples Using Single Metagenomic Next-Generation Sequencing Workflow. BMC Infect. Dis. 2018, 18, 537. [Google Scholar] [CrossRef]
- Leon, L.J.; Doyle, R.; Diez-Benavente, E.; Clark, T.G.; Klein, N.; Stanier, P.; Moore, G.E. Enrichment of Clinically Relevant Organisms in Spontaneous Preterm-Delivered Placentas and Reagent Contamination across All Clinical Groups in a Large Pregnancy Cohort in the United Kingdom. Appl. Environ. Microbiol. 2018, 84, e00483-18. [Google Scholar] [CrossRef]
- Qing, T.; Yu, Y.; Du, T.; Shi, L. MRNA Enrichment Protocols Determine the Quantification Characteristics of External RNA Spike-in Controls in RNA-Seq Studies. Sci. China Life Sci. 2013, 56, 134–142. [Google Scholar] [CrossRef][Green Version]
- Eisenhofer, R.; Minich, J.J.; Marotz, C.; Cooper, A.; Knight, R.; Weyrich, L.S. Contamination in Low Microbial Biomass Microbiome Studies: Issues and Recommendations. Trends Microbiol. 2019, 27, 105–117. [Google Scholar] [CrossRef]
- Matranga, C.B.; Andersen, K.G.; Winnicki, S.; Busby, M.; Gladden, A.D.; Tewhey, R.; Stremlau, M.; Berlin, A.; Gire, S.K.; England, E.; et al. Enhanced Methods for Unbiased Deep Sequencing of Lassa and Ebola RNA Viruses from Clinical and Biological Samples. Genome Biol. 2014, 15, 519. [Google Scholar] [CrossRef]
- Munro, S.A.; Lund, S.P.; Pine, P.S.; Binder, H.; Clevert, D.-A.; Conesa, A.; Dopazo, J.; Fasold, M.; Hochreiter, S.; Hong, H.; et al. Assessing Technical Performance in Differential Gene Expression Experiments with External Spike-in RNA Control Ratio Mixtures. Nat. Commun. 2014, 5, 5125. [Google Scholar] [CrossRef]
- Risso, D.; Ngai, J.; Speed, T.P.; Dudoit, S. Normalization of RNA-Seq Data Using Factor Analysis of Control Genes or Samples. Nat. Biotechnol. 2014, 32, 896–902. [Google Scholar] [CrossRef]
- Schlaberg, R.; Chiu, C.Y.; Miller, S.; Procop, G.W.; Weinstock, G. Validation of Metagenomic Next-Generation Sequencing Tests for Universal Pathogen Detection. Arch. Pathol. Lab. Med. 2017, 141, 776–786. [Google Scholar] [CrossRef]
- Zhou, Y.; Fernandez, S.; Yoon, I.-K.; Simasathien, S.; Watanaveeradej, V.; Yang, Y.; Marte-Salcedo, O.A.; Shuck-Lee, D.J.; Thomas, S.J.; Hang, J.; et al. Metagenomics Study of Viral Pathogens in Undiagnosed Respiratory Specimens and Identification of Human Enteroviruses at a Thailand Hospital. Am. J. Trop. Med. Hyg. 2016, 95, 663–669. [Google Scholar] [CrossRef]
- Capobianchi, M.R.; Giombini, E.; Rozera, G. Next-Generation Sequencing Technology in Clinical Virology. Clin. Microbiol. Infect. 2013, 19, 15–22. [Google Scholar] [CrossRef]
- Mostafa, H.H.; Fissel, J.A.; Fanelli, B.; Bergman, Y.; Gniazdowski, V.; Dadlani, M.; Carroll, K.C.; Colwell, R.R.; Simner, P.J. Metagenomic Next-Generation Sequencing of Nasopharyngeal Specimens Collected from Confirmed and Suspect COVID-19 Patients. mBio 2020, 11, e01969-20. [Google Scholar] [CrossRef]
- Qian, Y.-Y.; Wang, H.-Y.; Zhou, Y.; Zhang, H.-C.; Zhu, Y.-M.; Zhou, X.; Ying, Y.; Cui, P.; Wu, H.-L.; Zhang, W.-H.; et al. Improving Pulmonary Infection Diagnosis with Metagenomic Next Generation Sequencing. Front. Cell. Infect. Microbiol. 2021, 10, 567615. [Google Scholar] [CrossRef]
- Lipkin, W.I.; Firth, C. Viral Surveillance and Discovery. Curr. Opin. Virol. 2013, 3, 199–204. [Google Scholar] [CrossRef]
- del Campo, J.A.; Parra-Sánchez, M.; Figueruela, B.; García-Rey, S.; Quer, J.; Gregori, J.; Bernal, S.; Grande, L.; Palomares, J.C.; Romero-Gómez, M. Hepatitis C Virus Deep Sequencing for Sub-Genotype Identification in Mixed Infections: A Real-Life Experience. Int. J. Infect. Dis. 2018, 67, 114–117. [Google Scholar] [CrossRef]
- Thi Kha Tu, N.; Thi Thu Hong, N.; Thi Han Ny, N.; My Phuc, T.; Thi Thanh Tam, P.; van Doorn, H.R.; Dang Trung Nghia, H.; Thao Huong, D.; An Han, D.; Thi Thu Ha, L.; et al. The Virome of Acute Respiratory Diseases in Individuals at Risk of Zoonotic Infections. Viruses 2020, 12, 960. [Google Scholar] [CrossRef]
- Day, J.M.; Ballard, L.L.; Duke, M.V.; Scheffler, B.E.; Zsak, L. Metagenomic Analysis of the Turkey Gut RNA Virus Community. Virol. J. 2010, 7, 313. [Google Scholar] [CrossRef] [PubMed]
- Bishop-Lilly, K.A.; Turell, M.J.; Willner, K.M.; Butani, A.; Nolan, N.M.E.; Lentz, S.M.; Akmal, A.; Mateczun, A.; Brahmbhatt, T.N.; Sozhamannan, S.; et al. Arbovirus Detection in Insect Vectors by Rapid, High-Throughput Pyrosequencing. PLoS Negl. Trop. Dis. 2010, 4, e878. [Google Scholar] [CrossRef] [PubMed]
- Palacios, G.; Druce, J.; Du, L.; Tran, T.; Birch, C.; Briese, T.; Conlan, S.; Quan, P.-L.; Hui, J.; Marshall, J.; et al. A New Arenavirus in a Cluster of Fatal Transplant-Associated Diseases. N. Engl. J. Med. 2008, 358, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Briese, T.; Paweska, J.T.; McMullan, L.K.; Hutchison, S.K.; Street, C.; Palacios, G.; Khristova, M.L.; Weyer, J.; Swanepoel, R.; Egholm, M.; et al. Genetic Detection and Characterization of Lujo Virus, a New Hemorrhagic Fever–Associated Arenavirus from Southern Africa. PLoS Pathog. 2009, 5, e1000455. [Google Scholar] [CrossRef] [PubMed]
- Quan, P.-L.; Wagner, T.A.; Briese, T.; Torgerson, T.R.; Hornig, M.; Tashmukhamedova, A.; Firth, C.; Palacios, G.; Baisre-De-Leon, A.; Paddock, C.D.; et al. Astrovirus Encephalitis in Boy with X-Linked Agammaglobulinemia. Emerg. Infect. Dis. 2010, 16, 918–925. [Google Scholar] [CrossRef]
- Phan, T.G.; Li, L.; O’Ryan, M.G.; Cortes, H.; Mamani, N.; Bonkoungou, I.J.O.; Wang, C.; Leutenegger, C.M.; Delwart, E. A Third Gyrovirus Species in Human Faeces. J. Gen. Virol. 2012, 93, 1356–1361. [Google Scholar] [CrossRef]
- Yu, J.; Li, J.; Ao, Y.; Duan, Z. Detection of Novel Viruses in Porcine Fecal Samples from China. Virol. J. 2013, 10, 39. [Google Scholar] [CrossRef]
- Boros, Á.; Nemes, C.; Pankovics, P.; Kapusinszky, B.; Delwart, E.; Reuter, G. Identification and Complete Genome Characterization of a Novel Picornavirus in Turkey (Meleagris gallopavo). J. Gen. Virol. 2012, 93, 2171–2182. [Google Scholar] [CrossRef][Green Version]
- Honkavuori, K.S.; Briese, T.; Krauss, S.; Sanchez, M.D.; Jain, K.; Hutchison, S.K.; Webster, R.G.; Lipkin, W.I. Novel Coronavirus and Astrovirus in Delaware Bay Shorebirds. PLoS ONE 2014, 9, e93395. [Google Scholar] [CrossRef]
- Phan, T.G.; Vo, N.P.; Aronen, M.; Jartti, L.; Jartti, T.; Delwart, E. Novel Human Gammapapillomavirus Species in a Nasal Swab. Genome Announc. 2013, 1, e00022-13. [Google Scholar] [CrossRef]
- Reuter, G.; Boros, Á.; Delwart, E.; Pankovics, P. Novel Seadornavirus (Family Reoviridae) Related to Banna Virus in Europe. Arch. Virol. 2013, 158, 2163–2167. [Google Scholar] [CrossRef] [PubMed]
- Gibson, R.M.; Meyer, A.M.; Winner, D.; Archer, J.; Feyertag, F.; Ruiz-Mateos, E.; Leal, M.; Robertson, D.L.; Schmotzer, C.L.; Quiñones-Mateu, M.E. Sensitive Deep-Sequencing-Based HIV-1 Genotyping Assay to Simultaneously Determine Susceptibility to Protease, Reverse Transcriptase, Integrase, and Maturation Inhibitors, as Well as HIV-1 Coreceptor Tropism. Antimicrob. Agents Chemother. 2014, 58, 2167–2185. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Zhang, H.; Ma, H.; Liu, D.; Li, W.; Kang, Y.; Yang, R.; Wang, J.; He, G.; Xie, X.; et al. Deep Sequencing of Hepatitis B Virus Basal Core Promoter and Precore Mutants in HBeAg-Positive Chronic Hepatitis B Patients. Sci. Rep. 2015, 5, 17950. [Google Scholar] [CrossRef][Green Version]
- Gaspareto, K.V.; Ribeiro, R.M.; de Mello Malta, F.; Gomes-Gouvêa, M.S.; Muto, N.H.; Romano, C.M.; Mendes-Correa, M.C.; Carrilho, F.J.; Sabino, E.C.; Pinho, J.R.R. Resistance-Associated Variants in HCV Subtypes 1a and 1b Detected by Ion Torrent Sequencing Platform. Antivir. Ther. 2016, 21, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Nougairede, A.; Bichaud, L.; Thiberville, S.-D.; Ninove, L.; Zandotti, C.; de Lamballerie, X.; Brouqui, P.; Charrel, R.N. Isolation of Toscana Virus from the Cerebrospinal Fluid of a Man with Meningitis in Marseille, France, 2010. Vector-Borne Zoonotic Dis. 2013, 13, 685–688. [Google Scholar] [CrossRef]
- Anthony, S.J.; St. Leger, J.A.; Navarrete-Macias, I.; Nilson, E.; Sanchez-Leon, M.; Liang, E.; Seimon, T.; Jain, K.; Karesh, W.; Daszak, P.; et al. Identification of a Novel Cetacean Polyomavirus from a Common Dolphin (Delphinus delphis) with Tracheobronchitis. PLoS ONE 2013, 8, e68239. [Google Scholar] [CrossRef]
- Kvisgaard, L.K.; Hjulsager, C.K.; Fahnøe, U.; Breum, S.Ø.; Ait-Ali, T.; Larsen, L.E. A Fast and Robust Method for Full Genome Sequencing of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Type 1 and Type 2. J. Virol. Methods 2013, 193, 697–705. [Google Scholar] [CrossRef]
- Steyer, A.; Gutiérrez-Aguire, I.; Kolenc, M.; Koren, S.; Kutnjak, D.; Pokorn, M.; Poljšak-Prijatelj, M.; Rački, N.; Ravnikar, M.; Sagadin, M.; et al. High Similarity of Novel Orthoreovirus Detected in a Child Hospitalized with Acute Gastroenteritis to Mammalian Orthoreoviruses Found in Bats in Europe. J. Clin. Microbiol. 2013, 51, 3818–3825. [Google Scholar] [CrossRef]
- Lorusso, A.; Marcacci, M.; Ancora, M.; Mangone, I.; Leone, A.; Marini, V.; Cammà, C.; Savini, G. Complete Genome Sequence of Bluetongue Virus Serotype 1 Circulating in Italy, Obtained through a Fast Next-Generation Sequencing Protocol. Genome Announc. 2014, 2, e00093-14. [Google Scholar] [CrossRef]
- Ndze, V.N.; Esona, M.D.; Achidi, E.A.; Gonsu, K.H.; Dóró, R.; Marton, S.; Farkas, S.; Ngeng, M.B.; Ngu, A.F.; Obama-Abena, M.T.; et al. Full Genome Characterization of Human Rotavirus A Strains Isolated in Cameroon, 2010–2011: Diverse Combinations of the G and P Genes and Lack of Reassortment of the Backbone Genes. Infect. Genet. Evol. 2014, 28, 537–560. [Google Scholar] [CrossRef]
- Van den Hoecke, S.; Verhelst, J.; Vuylsteke, M.; Saelens, X. Analysis of the Genetic Diversity of Influenza A Viruses Using Next-Generation DNA Sequencing. BMC Genom. 2015, 16, 79. [Google Scholar] [CrossRef] [PubMed]
- Bzhalava, D.; Johansson, H.; Ekström, J.; Faust, H.; Möller, B.; Eklund, C.; Nordin, P.; Stenquist, B.; Paoli, J.; Persson, B.; et al. Unbiased Approach for Virus Detection in Skin Lesions. PLoS ONE 2013, 8, e65953. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Hu, C.; Zhang, D.; Tang, S.; Zhang, Z.; Kou, Z.; Fan, Z.; Bente, D.; Zeng, C.; Li, T. Metagenomic Profile of the Viral Communities in Rhipicephalus Spp. Ticks from Yunnan, China. PLoS ONE 2015, 10, e0121609. [Google Scholar] [CrossRef]
- Karlsson, O.E.; Larsson, J.; Hayer, J.; Berg, M.; Jacobson, M. The Intestinal Eukaryotic Virome in Healthy and Diarrhoeic Neonatal Piglets. PLoS ONE 2016, 11, e0151481. [Google Scholar] [CrossRef]
- Kluge, M.; Campos, F.S.; Tavares, M.; de Amorim, D.B.; Valdez, F.P.; Giongo, A.; Roehe, P.M.; Franco, A.C. Metagenomic Survey of Viral Diversity Obtained from Feces of Subantarctic and South American Fur Seals. PLoS ONE 2016, 11, e0151921. [Google Scholar] [CrossRef]
- Warwick-Dugdale, J.; Solonenko, N.; Moore, K.; Chittick, L.; Gregory, A.C.; Allen, M.J.; Sullivan, M.B.; Temperton, B. Long-Read Viral Metagenomics Captures Abundant and Microdiverse Viral Populations and Their Niche-Defining Genomic Islands. PeerJ 2019, 7, e6800. [Google Scholar] [CrossRef]
- Naveca, F.G.; Claro, I.; Giovanetti, M.; de Jesus, J.G.; Xavier, J.; Iani, F.C.d.M.; do Nascimento, V.A.; de Souza, V.C.; Silveira, P.P.; Lourenço, J.; et al. Genomic, Epidemiological and Digital Surveillance of Chikungunya Virus in the Brazilian Amazon. PLoS Negl. Trop. Dis. 2019, 13, e0007065. [Google Scholar] [CrossRef]
- Mohsin, H.; Asif, A.; Fatima, M.; Rehman, Y. Potential Role of Viral Metagenomics as a Surveillance Tool for the Early Detection of Emerging Novel Pathogens. Arch. Microbiol. 2021, 203, 865–872. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef]
- Posada-Cespedes, S.; Seifert, D.; Beerenwinkel, N. Recent Advances in Inferring Viral Diversity from High-Throughput Sequencing Data. Virus Res. 2017, 239, 17–32. [Google Scholar] [CrossRef]
- Rose, R.; Constantinides, B.; Tapinos, A.; Robertson, D.L.; Prosperi, M. Challenges in the Analysis of Viral Metagenomes. Virus Evol. 2016, 2, vew022. [Google Scholar] [CrossRef]
- Taş, N.; de Jong, A.E.; Li, Y.; Trubl, G.; Xue, Y.; Dove, N.C. Metagenomic Tools in Microbial Ecology Research. Curr. Opin. Biotechnol. 2021, 67, 184–191. [Google Scholar] [CrossRef]
- Nooij, S.; Schmitz, D.; Vennema, H.; Kroneman, A.; Koopmans, M.P.G. Overview of Virus Metagenomic Classification Methods and Their Biological Applications. Front. Microbiol. 2018, 9, 749. [Google Scholar] [CrossRef]
- Yang, C.; Chowdhury, D.; Zhang, Z.; Cheung, W.K.; Lu, A.; Bian, Z.; Zhang, L. A Review of Computational Tools for Generating Metagenome-Assembled Genomes from Metagenomic Sequencing Data. Comput. Struct. Biotechnol. J. 2021, 19, 6301–6314. [Google Scholar] [CrossRef]
- Kayani, M.u.R.; Huang, W.; Feng, R.; Chen, L. Genome-Resolved Metagenomics Using Environmental and Clinical Samples. Brief. Bioinform. 2021, 22, bbab030. [Google Scholar] [CrossRef]
- Sharma, D.; Priyadarshini, P.; Vrati, S. Unraveling the Web of Viroinformatics: Computational Tools and Databases in Virus Research. J. Virol. 2015, 89, 1489–1501. [Google Scholar] [CrossRef]
- Metsky, H.C.; Siddle, K.J.; Gladden-Young, A.; Qu, J.; Yang, D.K.; Brehio, P.; Goldfarb, A.; Piantadosi, A.; Wohl, S.; Carter, A.; et al. Capturing Sequence Diversity in Metagenomes with Comprehensive and Scalable Probe Design. Nat. Biotechnol. 2019, 37, 160–168. [Google Scholar] [CrossRef]
- Lorenzi, H. Viral Metagenome Annotation Pipeline. In Encyclopedia of Metagenomics; Springer: New York, NY, USA, 2013; pp. 1–12. [Google Scholar]
- Wommack, K.E.; Bhavsar, J.; Polson, S.W.; Chen, J.; Dumas, M.; Srinivasiah, S.; Furman, M.; Jamindar, S.; Nasko, D.J. VIROME: A Standard Operating Procedure for Analysis of Viral Metagenome Sequences. Stand. Genom. Sci. 2012, 6, 427–439. [Google Scholar] [CrossRef]
- Roux, S.; Tournayre, J.; Mahul, A.; Debroas, D.; Enault, F. Metavir 2: New Tools for Viral Metagenome Comparison and Assembled Virome Analysis. BMC Bioinform. 2014, 15, 76. [Google Scholar] [CrossRef]
- Maabar, M.; Davison, A.J.; Vučak, M.; Thorburn, F.; Murcia, P.R.; Gunson, R.; Palmarini, M.; Hughes, J. DisCVR: Rapid Viral Diagnosis from High-Throughput Sequencing Data. Virus Evol. 2019, 5, vez033. [Google Scholar] [CrossRef]
- Naccache, S.N.; Federman, S.; Veeraraghavan, N.; Zaharia, M.; Lee, D.; Samayoa, E.; Bouquet, J.; Greninger, A.L.; Luk, K.-C.; Enge, B.; et al. A Cloud-Compatible Bioinformatics Pipeline for Ultrarapid Pathogen Identification from next-Generation Sequencing of Clinical Samples. Genome Res. 2014, 24, 1180–1192. [Google Scholar] [CrossRef]
- Kalantar, K.L.; Carvalho, T.; de Bourcy, C.F.A.; Dimitrov, B.; Dingle, G.; Egger, R.; Han, J.; Holmes, O.B.; Juan, Y.-F.; King, R.; et al. IDseq—An Open Source Cloud-Based Pipeline and Analysis Service for Metagenomic Pathogen Detection and Monitoring. Gigascience 2020, 9, giaa111. [Google Scholar] [CrossRef]
- Hasan, N.A.; Young, B.A.; Minard-Smith, A.T.; Saeed, K.; Li, H.; Heizer, E.M.; McMillan, N.J.; Isom, R.; Abdullah, A.S.; Bornman, D.M.; et al. Microbial Community Profiling of Human Saliva Using Shotgun Metagenomic Sequencing. PLoS ONE 2014, 9, e97699. [Google Scholar] [CrossRef]
- Ponnusamy, D.; Kozlova, E.V.; Sha, J.; Erova, T.E.; Azar, S.R.; Fitts, E.C.; Kirtley, M.L.; Tiner, B.L.; Andersson, J.A.; Grim, C.J.; et al. Cross-Talk among Flesh-Eating Aeromonas Hydrophila Strains in Mixed Infection Leading to Necrotizing Fasciitis. Proc. Natl. Acad. Sci. USA 2016, 113, 722–727. [Google Scholar] [CrossRef]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN Analysis of Metagenomic Data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef]
- Breitwieser, F.P.; Salzberg, S.L. Pavian: Interactive Analysis of Metagenomics Data for Microbiome Studies and Pathogen Identification. Bioinformatics 2020, 36, 1303–1304. [Google Scholar] [CrossRef]
- Pedersen, T.L.; Nookaew, I.; Wayne Ussery, D.; Månsson, M. PanViz: Interactive Visualization of the Structure of Functionally Annotated Pangenomes. Bioinformatics 2017, 33, 1081–1082. [Google Scholar] [CrossRef]
- Wagner, J.; Chelaru, F.; Kancherla, J.; Paulson, J.N.; Zhang, A.; Felix, V.; Mahurkar, A.; Elmqvist, N.; Corrada Bravo, H. Metaviz: Interactive Statistical and Visual Analysis of Metagenomic Data. Nucleic Acids Res. 2018, 46, 2777–2787. [Google Scholar] [CrossRef]
- Eren, A.M.; Esen, Ö.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An Advanced Analysis and Visualization Platform for ‘omics Data. PeerJ 2015, 3, e1319. [Google Scholar] [CrossRef]
- Skewes-Cox, P.; Sharpton, T.J.; Pollard, K.S.; DeRisi, J.L. Profile Hidden Markov Models for the Detection of Viruses within Metagenomic Sequence Data. PLoS ONE 2014, 9, e105067. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Sekizuka, T.; Kuroda, M. VirusTAP: Viral Genome-Targeted Assembly Pipeline. Front. Microbiol. 2016, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Kramna, L.; Autio, R.; Hyöty, H.; Nykter, M.; Cinek, O. Vipie: Web Pipeline for Parallel Characterization of Viral Populations from Multiple NGS Samples. BMC Genom. 2017, 18, 378. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-H.; Liao, Y.-C. DrVM: A New Tool for Efficient Genome Assembly of Known Eukaryotic Viruses from Metagenomes. Gigascience 2017, 6, gix003. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Chilton, J.; Coraor, N.; Coppens, F.; Eguinoa, I.; Gladman, S.; Grüning, B.; et al. No More Business as Usual: Agile and Effective Responses to Emerging Pathogen Threats Require Open Data and Open Analytics. PLoS Pathog. 2020, 16, e1008643. [Google Scholar] [CrossRef]
- Lecuit, M.; Eloit, M. The Diagnosis of Infectious Diseases by Whole Genome next Generation Sequencing: A New Era Is Opening. Front. Cell. Infect. Microbiol. 2014, 4, 25. [Google Scholar] [CrossRef]
- Prachayangprecha, S.; Schapendonk, C.M.E.; Koopmans, M.P.; Osterhaus, A.D.M.E.; Schürch, A.C.; Pas, S.D.; van der Eijk, A.A.; Poovorawan, Y.; Haagmans, B.L.; Smits, S.L. Exploring the Potential of Next-Generation Sequencing in Detection of Respiratory Viruses. J. Clin. Microbiol. 2014, 52, 3722–3730. [Google Scholar] [CrossRef]
- Baillie, G.J.; Galiano, M.; Agapow, P.-M.; Myers, R.; Chiam, R.; Gall, A.; Palser, A.L.; Watson, S.J.; Hedge, J.; Underwood, A.; et al. Evolutionary Dynamics of Local Pandemic H1N1/2009 Influenza Virus Lineages Revealed by Whole-Genome Analysis. J. Virol. 2012, 86, 11–18. [Google Scholar] [CrossRef]
- Li, D.; Li, Z.; Zhou, Z.; Li, Z.; Qu, X.; Xu, P.; Zhou, P.; Bo, X.; Ni, M. Direct Next-Generation Sequencing of Virus-Human Mixed Samples without Pretreatment Is Favorable to Recover Virus Genome. Biol. Direct 2016, 11, 3. [Google Scholar] [CrossRef][Green Version]



{kind=link}
{kind=link}
| # | Software | Description | References |
|---|---|---|---|
| Processing of Sequencing Data | |||
| 1 | Trimmomatic | It is a command line tool that can be used to trim and crop Illumina (FASTQ) data as well as to remove adapters | http://www.usadellab.org/cms/?page=trimmomatic (accessed on 14 February 2022) |
| 2 | PRINSEQ | It is a tool for quality control of metagenomic sequence data, includes functions for statistics, for trimming, filtering, and data reformatting | https://bioinformaticshome.com/tools/rna-seq/descriptions/PRINSEQ.html#:~:text=PRINSEQ%20is%20a%20tool%20for,quality%20measures%2C%20and%20tag%20sequences (accessed on 14 February 2022). |
| 3 | Cutadapt | Finds and removes adapter sequences, primers, poly-A tails and other types of unwanted sequence from high-throughput sequencing reads | https://cutadapt.readthedocs.io/en/stable/ (accessed on 14 February 2022) |
| 4 | MEGAN | For analysis of large metagenomic datasets. The set of DNA reads compared against databases of known sequences | https://bio.tools/megan (accessed on 14 February 2022) |
| Virus genome analysis | |||
| 5 | Metavir 2 | For a comprehensive analysis of assembled virome sequences | https://www.cd-genomics.com/bioinformatics-analysis-of-viral-metagenomic-sequencing.html (accessed on 14 February 2022) |
| 6 | MetaGeneAnnotator | It is a gene-finding program for prokaryote and phage | https://mybiosoftware.com/tag/metageneannotator (accessed on 14 February 2022) |
| 7 | VIP | For metagenomic identification of viral pathogens | https://github.com/keylabivdc/VIP (accessed on 14 February 2022) |
| 8 | VirusSeker | For eukaryotic virus discovery and composition analysis | https://mybiosoftware.com/tag/virusseeker (accessed on 14 February 2022) |
| 9 | VirusTAP | It is analysis tool for the viral genome | https://gph.niid.go.jp/cgi-bin/virustap/index.cgi (accessed on 14 February 2022) |
| Taxonomic classification | |||
| 10 | DisCVR | For detection of known human viruses in clinical samples from high-throughput sequencing | https://bioinformatics.cvr.ac.uk/software/discvr/ (accessed on 14 February 2022) |
| 11 | KRAKEN | For assigning taxonomic labels to short DNA sequences, usually obtained through metagenomic studies | https://ccb.jhu.edu/software/kraken/ (accessed on 14 February 2022) |
| 12 | Bracken | It is a highly accurate statistical method for species of DNA. Bracken uses the taxonomy labels assigned by Kraken | https://ccb.jhu.edu/software/bracken/ (accessed on 14 February 2022) |
| 13 | Centrifuge | It is a microbial classification engine that enables rapid, accurate, and sensitive labeling of reads and quantification of species | https://ccb.jhu.edu/software/centrifuge/manual.shtml (accessed on 14 February 2022) |
| 14 | CLARK | For classification of short metagenomics reads at the genus/species using discriminative k-mers | http://clark.cs.ucr.edu/ (accessed on 14 February 2022) |
| 15 | VIROME | For classification of predicted open-reading frames (ORFs) from viral metagenomes | http://virome.dbi.udel.edu/ (accessed on 14 February 2022) |
| 16 | Taxonomer | For assigning taxonomy to sequencing reads from both clinical and environmental samples | https://taxonomer.iobio.io/#:~:text=Taxonomer%20is%20a%20kmer%2Dbased,meaningful%20timeframe%20(i.e.,%20minutes) (accessed on 14 February 2022) |
| Visualization tools | |||
| 17 | Pavian | For exploring metagenomics classification results, with a special focus on infectious disease diagnosis. Analyze, display, and transform results from the Kraken and Centrifuge | https://ccb.jhu.edu/software/pavian/ (accessed on 16 February 2022) |
| 18 | Krona | It is an interactive visualization tool for exploring the composition of metagenomes | https://sourceforge.net/p/krona/home/krona/?version=27 (accessed on 16 February 2022) |
| 19 | PanViz | Visualization tool for investigating and understanding comparative microbial genomics data | https://github.com/thomasp85/PanViz (accessed on 16 February 2022) |
| 20 | MetaViz | For interactive exploratory data analysis of annotated microbiome taxonomic community profiles | https://mybiosoftware.com/tag/metaviz (accessed on 16 February 2022) |
| 21 | Anvi’o | It is an analysis and visualization platform that offers automated and human-guided characterization of microbial genomes in metagenomic assemblies | https://anvio.org/ (accessed on 16 February 2022) |
| 22 | Geneious | It is a DNA, RNA, and protein sequence alignment, assembly, and analysis software platform, integrating bioinformatics and molecular biology tools | https://www.geneious.com/ (accessed on 16 February 2022) |
| 23 | CLC bio | For bioinformatics analysis with graphical interface for building, managing and deploying analysis workflows | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-clc-genomics-workbench/ (accessed on 16 February 2022) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandybayev, N.; Beloussov, V.; Strochkov, V.; Solomadin, M.; Granica, J.; Yegorov, S. Next Generation Sequencing Approaches to Characterize the Respiratory Tract Virome. Microorganisms 2022, 10, 2327. https://doi.org/10.3390/microorganisms10122327
Sandybayev N, Beloussov V, Strochkov V, Solomadin M, Granica J, Yegorov S. Next Generation Sequencing Approaches to Characterize the Respiratory Tract Virome. Microorganisms. 2022; 10(12):2327. https://doi.org/10.3390/microorganisms10122327
Chicago/Turabian StyleSandybayev, Nurlan, Vyacheslav Beloussov, Vitaliy Strochkov, Maxim Solomadin, Joanna Granica, and Sergey Yegorov. 2022. "Next Generation Sequencing Approaches to Characterize the Respiratory Tract Virome" Microorganisms 10, no. 12: 2327. https://doi.org/10.3390/microorganisms10122327
APA StyleSandybayev, N., Beloussov, V., Strochkov, V., Solomadin, M., Granica, J., & Yegorov, S. (2022). Next Generation Sequencing Approaches to Characterize the Respiratory Tract Virome. Microorganisms, 10(12), 2327. https://doi.org/10.3390/microorganisms10122327