
Systems Biology: New Insight into Antibiotic Resistance


Abstract
1. Introduction
2. Antibiotic Resistance: A Complex and Multilevel Problem
3. Mechanisms of Action and Resistance Acquisition
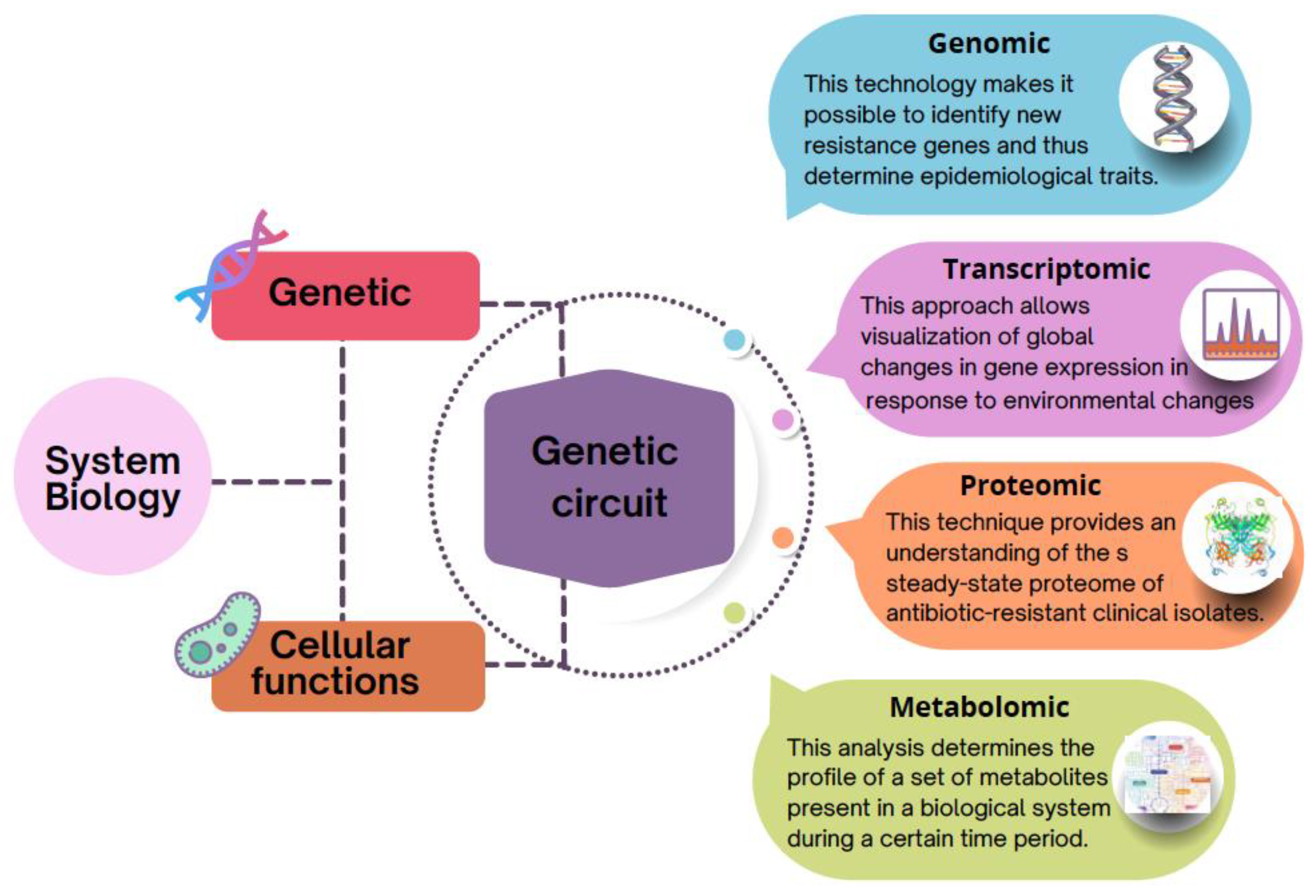
4. Systems Level: The Increasing Use of the Post-Genomic Approach to Understanding Antibiotic Resistance
4.1. Genomics: First Step to Identify Antibiotic Resistance Genes (ARG)
4.2. Emerging Discoveries through Transcriptomics Approach
4.3. Recent Trends in Proteomics
4.4. Accelerated Growth in the Use of Metabolomics
4.5. Metabolic Models to Expand the Comprehension of Mechanisms Associated with Antibiotic Resistance
5. Limitation of the Use of Systems Biology
6. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Laws, M.; Shaaban, A.; Rahman, K.M. Antibiotic Resistance Breakers: Current Approaches and Future Directions. FEMS Microbiol. Rev. 2019, 43, 490–516. [Google Scholar] [CrossRef] [PubMed]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How Antibiotics Kill Bacteria: From Targets to Networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Alcalde-Rico, M.; Olivares-Pacheco, J.; Halliday, N.; Cámara, M.; Martínez, J.L. The Impaired Quorum Sensing Response of Pseudomonas aeruginosa MexAB-OprM Efflux Pump Overexpressing Mutants Is Not Due to Non-Physiological Efflux of 3-Oxo-C12-HSL. Environ. Microbiol 2020, 22, 5167–5188. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Banerjee, S.K.; Talukdar, N.C.; Yadav, A.K. Post-Translational Modification Crosstalk and Hotspots in Sirtuin Interactors Implicated in Cardiovascular Diseases. Front. Genet. 2020, 11, 356. [Google Scholar] [CrossRef] [PubMed]
- Adela, R.; Reddy, P.N.C.; Ghosh, T.S.; Aggarwal, S.; Yadav, A.K.; Das, B.; Banerjee, S.K. Serum Protein Signature of Coronary Artery Disease in Type 2 Diabetes Mellitus. J. Transl. Med. 2019, 17, 17. [Google Scholar] [CrossRef]
- Yadav, A.K.; Banerjee, S.K.; Das, B.; Chaudhary, K. Editorial: Systems Biology and Omics Approaches for Understanding Complex Disease Biology. Front. Genet. 2022, 13, 896818. [Google Scholar] [CrossRef]
- Gil-Gil, T.; Ochoa-Sánchez, L.E.; Baquero, F.; Martínez, J.L. Antibiotic resistance: Time of synthesis in a post-genomic age. Comput. Struct. Biotechnol. J. 2021, 19, 3110–3124. [Google Scholar] [CrossRef]
- Sukhum, K.V.; Diorio-Toth, L.; Dantas, G. Genomic and Metagenomic Approaches for Predictive Surveillance of Emerging Pathogens and Antibiotic Resistance. Clin. Pharm. 2019, 106, 512–524. [Google Scholar] [CrossRef]
- Santiago-Rodriguez, T.M.; Hollister, E.B. Multi ‘omic Data Integration: A Review of Concepts, Considerations, and Approaches. Semin. Perinatol. 2021, 45, 151456. [Google Scholar] [CrossRef]
- Ndagi, U.; Falaki, A.A.; Abdullahi, M.; Lawal, M.M.; Soliman, M.E. Antibiotic Resistance: Bioinformatics-Based Understanding as a Functional Strategy for Drug Design. RSC Adv. 2020, 10, 18451–18468. [Google Scholar] [CrossRef]
- Morehead, M.S.; Scarbrough, C. Emergence of Global Antibiotic Resistance. Prim. Care-Clin. Off. Pract. 2018, 45, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Hwang, A.Y.; Gums, J.G. The Emergence and Evolution of Antimicrobial Resistance: Impact on a Global Scale. Bioorg. Med. Chem. 2016, 24, 6440–6445. [Google Scholar] [CrossRef] [PubMed]
- Fair, R.J.; Tor, Y. Antibiotics and Bacterial Resistance in the 21st Century. Perspect. Med. Chem. 2014, 6, 25–64. [Google Scholar] [CrossRef] [PubMed]
- Brazas, M.D.; Hancock, R.E.W. Using Microarray Gene Signatures to Elucidate Mechanisms of Antibiotic Action and Resistance. Drug Discov. Today 2005, 10, 1245–1252. [Google Scholar] [CrossRef]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4, 464–473. [Google Scholar] [CrossRef]
- Alanis, A.J. Resistance to Antibiotics: Are We in the Post-Antibiotic Era? Arch. Med. Res. 2005, 36, 697–705. [Google Scholar] [CrossRef]
- Author, R.; Fleischmann, R.D.; Adams, M.D.; White, O.; Clayton, R.A.; Kirkness, E.F.; Kerlavage, A.R.; Bult, C.J.; Tomb, J.-F.; Dougherty, B.A.; et al. Whole-Genome Random Sequencing and Assembly of Haemophilus Influenzae. Science 1995, 269, 496–512. [Google Scholar]
- Sandoval-Motta, S.; Aldana, M. Adaptive resistance to antibiotics in bacteria: A systems biology perspective. WIREs Syst. Biol. Med. 2016, 8, 253–267. [Google Scholar] [CrossRef]
- Turanli, B.; Altay, O.; Borén, J.; Turkez, H.; Nielsen, J.; Uhlen, M.; Arga, K.Y.; Mardinoglu, A. Systems biology based drug repositioning for development of cancer therapy. Semin. Cancer Biol. 2019, 68, 47–58. [Google Scholar] [CrossRef]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.M. ARG-Annot, a New Bioinformatic Tool to Discover Antibiotic Resistance Genes in Bacterial Genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Feldgarden, M.; Brover, V.; Haft, D.H.; Prasad, A.B.; Slotta, D.J.; Tolstoy, I.; Tyson, G.H.; Zhao, S.; Hsu, C.-H.; McDermott, P.F.; et al. Validating the AMRFinder Tool and Resistance Gene Database by Using Antimicrobial Resistance Genotype-Phenotype Correlations in a Collection of Isolates. Antimicrob. Agents Chemother. 2019, 63, e00483-19. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Allesøe, R.; Joensen, K.G.; Cavaco, L.M.; Lund, O.; Aarestrup, F.M. PointFinder: A Novel Web Tool for WGS-Based Detection of Antimicrobial Resistance Associated with Chromosomal Point Mutations in Bacterial Pathogens. J. Antimicrob. Chemother. 2017, 72, 2764–2768. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular Framework for Processing, Visualizing, and Analyzing Mass Spectrometry-Based Molecular Profile Data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the Gap between Raw Spectra and Functional Insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Shen, X.; Zhu, Z.J. MetFlow: An Interactive and Integrated Workflow for Metabolomics Data Cleaning and Differential Metabolite Discovery. Bioinformatics 2019, 35, 2870–2872. [Google Scholar] [CrossRef]
- Zhou, G.; Xia, J. OmicsNet: A Web-Based Tool for Creation and Visual Analysis of Biological Networks in 3D Space. Nucleic Acids Res. 2018, 46, W514–W522. [Google Scholar] [CrossRef] [PubMed]
- Hernández-De-Diego, R.; Tarazona, S.; Martínez-Mira, C.; Balzano-Nogueira, L.; Furió-Tarí, P.; Pappas, G.J.; Conesa, A. PaintOmics 3: A Web Resource for the Pathway Analysis and Visualization of Multi-Omics Data. Nucleic Acids Res. 2018, 46, W503–W509. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision Weights Unlock Linear Model Analysis Tools for RNA-Seq Read Counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-A Python Framework to Work with High-Throughput Sequencing Data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Schmid, M.W.; Grossniklaus, U. Rcount: Simple and Flexible RNA-Seq Read Counting. Bioinformatics 2015, 31, 436–437. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential Gene and Transcript Expression Analysis of RNA-Seq Experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Karp, P.D.; Paley, S.M.; Midford, P.E.; Krummenacker, M.; Billington, R.; Kothari, A.; Ong, W.K.; Subhraveti, P.; Keseler, I.M.; Caspi, R. Pathway Tools Version 24.0: Integrated Software for Pathway/Genome Informatics and Systems Biology. arXiv 2015, arXiv:1510.03964. [Google Scholar]
- Garcia-Albornoz, M.; Thankaswamy-Kosalai, S.; Nilsson, A.; Väremo, L.; Nookaew, I.; Nielsen, J. BioMet Toolbox 2.0: Genome-wide analysis of metabolism and omics data. Nucleic Acids Res. 2014, 42, W175–W181. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for Taxonomy-Based Analysis of Pathways and Genomes. Nucleic Acids Res. 2022, gkac963. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium. The Gene Ontology Resource: Enriching a GOld Mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef] [PubMed]
- Karp, P.D.; Billington, R.; Caspi, R.; A Fulcher, C.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; E Midford, P.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. Briefings Bioinform. 2017, 20, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [PubMed]
- Sertbas, M.; Ulgen, K.O. Genome-Scale Metabolic Modeling for Unraveling Molecular Mechanisms of High Threat Pathogens. Front. Cell Dev. Biol. 2020, 8, 566702. [Google Scholar] [CrossRef]
- Hendriksen, R.S.; Bortolaia, V.; Tate, H.; Tyson, G.H.; Aarestrup, F.; McDermott, P.F. Using Genomics to Track Global Antimicrobial Resistance. Front. Public Health 2019, 7, 242. [Google Scholar] [CrossRef]
- Heinemann, M.; Kümmel, A.; Ruinatscha, R.; Panke, S. In silico genome-scale reconstruction and validation of the Staphylococcus aureus metabolic network. Biotechnol. Bioeng. 2005, 92, 850–864. [Google Scholar] [CrossRef]
- Wieser, A.; Schneider, L.; Jung, J.; Schubert, S. MALDI-TOF MS in microbiological diagnostics—identification of microorganisms and beyond (mini review). Appl. Microbiol. Biotechnol. 2011, 93, 965–974. [Google Scholar] [CrossRef]
- Reller, L.B.; Weinstein, M.; Jorgensen, J.H.; Ferraro, M.J. Antimicrobial Susceptibility Testing: A Review of General Principles and Contemporary Practices. Clin. Infect. Dis. 2009, 49, 1749–1755. [Google Scholar] [CrossRef]
- Didelot, X.; Bowden, R.; Wilson, D.J.; Peto, T.E.A.; Crook, D.W. Transforming clinical microbiology with bacterial genome sequencing. Nat. Rev. Genet. 2012, 13, 601–612. [Google Scholar] [CrossRef]
- Hwang, S.M.; Cho, H.W.; Kim, T.Y.; Park, J.S.; Jung, J.; Song, K.-H.; Lee, H.; Kim, E.S.; Bin Kim, H.; Park, K.U. Whole-Genome Sequencing for Investigating a Health Care-Associated Outbreak of Carbapenem-Resistant Acinetobacter baumannii. Diagnostics 2021, 11, 201. [Google Scholar] [CrossRef]
- Grad, Y.H.; Kirkcaldy, R.D.; Trees, D.; Dordel, J.; Harris, S.R.; Goldstein, E.; Weinstock, H.; Parkhill, J.; Hanage, W.P.; Bentley, S.; et al. Genomic epidemiology of Neisseria gonorrhoeae with reduced susceptibility to cefixime in the USA: A retrospective observational study. Lancet Infect. Dis. 2014, 14, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Grad, Y.H.; Harris, S.R.; Kirkcaldy, R.D.; Green, A.G.; Marks, D.S.; Bentley, S.D.; Trees, D.; Lipsitch, M. Genomic Epidemiology of Gonococcal Resistance to Extended-Spectrum Cephalosporins, Macrolides, and Fluoroquinolones in the United States, 2000–2013. J. Infect. Dis. 2016, 214, 1579–1587. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.T.; Vítor, J.M.B.; Santos, A.; Oleastro, M.; Vale, F.F. Trends in Helicobacter pylori resistance to clarithromycin: From phenotypic to genomic approaches. Microb. Genom. 2020, 6, e000344. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Liu, S.; Ying, Y.; Xu, L.; Liu, Y.; Jin, J.; Ying, J.; Lu, J.; Lin, X.; Li, K.; et al. Genomic and functional characterization of fecal sample strains of Proteus cibarius carrying two floR antibiotic resistance genes and a multiresistance plasmid-encoded cfr gene. Comp. Immunol. Microbiol. Infect. Dis. 2020, 69, 101427. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhang, X.; Liang, J.; Li, Q.; Lin, H.; Lin, C.; Liu, H.; Zhou, D.; Lu, W.; Sun, Z.; et al. Characterization of florfenicol resistance genes in the coagulase-negative Staphylococcus (CoNS) isolates and genomic features of a multidrug-resistant Staphylococcus lentus strain H29. Antimicrob. Resist. Infect. Control 2021, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, R.; Li, W.; Ma, J.; Lin, H. Genomic Insights into the Antibiotic Resistance Pattern of the Tetracycline-Degrading Bacterium, Arthrobacter nicotianae OTC-16. Sci. Rep. 2021, 11, 15638. [Google Scholar] [CrossRef] [PubMed]
- Boiko, I.; Golparian, D.; Jacobsson, S.; Krynytska, I.; Frankenberg, A.; Shevchenko, T.; Unemo, M. Genomic Epidemiology and Antimicrobial Resistance Determinants of Neisseria Gonorrhoeae Isolates from Ukraine, 2013–2018. APMIS 2020, 128, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Rokney, A.; Valinsky, L.; Vranckx, K.; Feldman, N.; Agmon, V.; Moran-Gilad, J.; Weinberger, M. WGS-Based Prediction and Analysis of Antimicrobial Resistance in Campylobacter jejuni Isolates From Israel. Front. Cell Infect. Microbiol. 2020, 10, 365. [Google Scholar] [CrossRef]
- D’Onofrio, V.; Conzemius, R.; Varda-Brkić, D.; Bogdan, M.; Grisold, A.; Gyssens, I.C.; Bedenić, B.; Barišić, I. Epidemiology of Colistin-Resistant, Carbapenemase-Producing Enterobacteriaceae and Acinetobacter baumannii in Croatia. Infect. Genet. Evol. 2020, 81, 104263. [Google Scholar] [CrossRef]
- Lee, T.; Pang, S.; Stegger, M.; Sahibzada, S.; Abraham, S.; Daley, D.; Coombs, G.; on behalf of the Australian Group on Antimicrobial Resistance. A three-year whole genome sequencing perspective of Enterococcus faecium sepsis in Australia. PLoS ONE 2020, 15, e0228781. [Google Scholar] [CrossRef]
- Butin, M.; Martins-Simões, P.; Pichon, B.; Leyssene, D.; Bordes-Couecou, S.; Meugnier, H.; Rouard, C.; Lemaitre, N.; Schramm, F.; Kearns, A.; et al. Emergence and Dissemination of a Linezolid-Resistant Staphylococcus Capitis Clone in Europe. J. Antimicrob. Chemother. 2017, 72, 1014–1020. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, J.; Vougas, K.; Shah, A.; Shah, H.; Misra, R.; Mkrtchyan, H.V. Comparative Proteomic Profiling of Methicillin-Susceptible and Resistant Staphylococcus Aureus. Proteomics 2020, 20, e1900221. [Google Scholar] [CrossRef]
- Ranjitkar, S.; Jones, A.K.; Mostafavi, M.; Zwirko, Z.; Iartchouk, O.; Barnes, S.W.; Walker, J.R.; Willis, T.W.; Lee, P.S.; Dean, C.R. Target (MexB)-and Efflux-Based Mechanisms Decreasing the Effectiveness of the Efflux Pump Inhibitor D13-9001 in Pseudomonas Aeruginosa PAO1: Uncovering a New Role for MexMN-OprM in Efflux of-Lactams and a Novel Regulatory Circuit (MmnRS) Controlling MexMN Expression. ASM J. 2019, 63, e01718-18. [Google Scholar] [CrossRef]
- Li, L.; Li, R.; Qi, C.; Gao, H.; Wei, Q.; Tan, L.; Sun, F. Mechanisms of Polymyxin Resistance Induced by Salmonella Typhimurium in Vitro. Vet. Microbiol. 2021, 257, 109063. [Google Scholar] [CrossRef] [PubMed]
- Wang-Kan, X.; A Blair, J.M.; Chirullo, B.; Betts, J.; la Ragione, R.M.; Ivens, A.; Ricci, V.; Opperman, T.J.; Piddock, L.J.V.; Wang-Kan, C.X.; et al. Lack of AcrB Efflux Function Confers Loss of Virulence on Salmonella Enterica Serovar Typhimurium. ASM J. 2017, 8, 968–985. [Google Scholar] [CrossRef]
- Nghiem, M.N.; Nguyen, V.T.; Jeung, E.B.; Vo, T.T.B. Alternate Antimicrobial Resistance Genes in Multidrug Resistant Salmonella spp. Isolated from Retail Meats in Vietnam Using RNA-Sequencing Analysis. J. Food Saf. 2019, 39, e12707. [Google Scholar] [CrossRef]
- Gu, Y.; Huang, L.; Wu, C.; Huang, J.; Hao, H.; Yuan, Z.; Cheng, G. The Evolution of Fluoroquinolone Resistance in Salmonella under Exposure to Sub-Inhibitory Concentration of Enrofloxacin. Int. J. Mol. Sci. 2021, 22, 12218. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, D.; Natarajan, J. RNA-Seq Analysis Reveals Resistome Genes and Signalling Pathway Associated with Vancomycin-Intermediate Staphylococcus aureus. Indian J. Med. Microbiol. 2019, 37, 173–185. [Google Scholar] [CrossRef]
- Wang, H.; Kraus, F.; Popella, P.; Baykal, A.; Guttroff, C.; François, P.; Sass, P.; Plietker, B.; Götz, F. The Polycyclic Polyprenylated Acylphloroglucinol Antibiotic PPAP 23 Targets the Membrane and Iron Metabolism in Staphylococcus aureus. Front. Microbiol. 2019, 10, 14. [Google Scholar] [CrossRef]
- Cho, H.; Misra, R. Mutational Activation of Antibiotic-Resistant Mechanisms in the Absence of Major Drug Efflux Systems of Escherichia coli. J. Bacteriol. 2021, 203, e0010921. [Google Scholar] [CrossRef] [PubMed]
- Alkasir, R.; Ma, Y.; Liu, F.; Li, J.; Lv, N.; Xue, Y.; Hu, Y.; Zhu, B. Characterization and Transcriptome Analysis of Acinetobacter Baumannii Persister Cells. Microb. Drug Resist. 2018, 24, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Kröger, C.; MacKenzie, K.D.; Alshabib, E.Y.; Kirzinger, M.W.B.; Suchan, D.M.; Chao, T.C.; Akulova, V.; Miranda-CasoLuengo, A.A.; Monzon, V.A.; Conway, T.; et al. The Primary Transcriptome, Small RNAs and Regulation of Antimicrobial Resistance in Acinetobacter Baumannii ATCC 17978. Nucleic Acids Res. 2018, 46, 9684–9698. [Google Scholar] [CrossRef] [PubMed]
- Mack, S.G.; Turner, R.L.; Dwyer, D.J. Achieving a Predictive Understanding of Antimicrobial Stress Physiology through Systems Biology. Trends Microbiol. 2018, 26, 296–312. [Google Scholar] [CrossRef]
- Florio, W.; Baldeschi, L.; Rizzato, C.; Tavanti, A.; Ghelardi, E.; Lupetti, A. Detection of Antibiotic-Resistance by MALDI-TOF Mass Spectrometry: An Expanding Area. Front. Cell. Infect. Microbiol. 2020, 10, 572909. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Gopinath, K.; Sharma, P.; Bisht, D.; Sharma, P.; Singh, N.; Singh, S. Comparative Proteomic Analysis of Sequential Isolates of Mycobacterium Tuberculosis from a Patient with Pulmonary Tuberculosis Turning from Drug Sensitive to Multidrug Resistant. Indian J. Med. Res. 2015, 141, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.J.; Ma, C.J.; Kim, J.C.; Ahn, J. Proteomics-Based Discrimination of Differentially Expressed Proteins in Antibiotic-Sensitive and Antibiotic-Resistant Salmonella typhimurium, Klebsiella pneumoniae, and Staphylococcus aureus. Arch. Microbiol. 2019, 201, 1259–1275. [Google Scholar] [CrossRef]
- Kittisenachai, S.; Rojpibulstit, P.; Vilaichone, R.K.; Gamnarai, P.; Phaonakrop, N.; Suealek, N. FBPAII and RpoBC, the Two Novel Secreted Proteins Identified by the Proteomic Approach from a Comparative Study between Antibiotic-Sensitive and Antibiotic-Resistant Helicobacter Pylori-Associated Gastritis Strains. Infect. Immun. 2021, 89, e00053-21. [Google Scholar] [CrossRef]
- Foudraine, D.E.; Strepis, N.; Stingl, C.; ten Kate, M.T.; Verbon, A.; Klaassen, C.H.W.; Goessens, W.H.F.; Luider, T.M.; Dekker, L.J.M. Exploring Antimicrobial Resistance to Beta-Lactams, Aminoglycosides and Fluoroquinolones in E. Coli and K. Pneumoniae Using Proteogenomics. Sci. Rep. 2021, 11, 12472. [Google Scholar] [CrossRef]
- Li, W.; Wang, G.; Zhang, S.; Fu, Y.; Jiang, Y.; Yang, X.; Lin, X. An Integrated Quantitative Proteomic and Metabolomics Approach to Reveal the Negative Regulation Mechanism of LamB in Antibiotics Resistance. J. Proteom. 2019, 194, 148–159. [Google Scholar] [CrossRef]
- Kok, M.; Maton, L.; van der Peet, M.; Hankemeier, T.; van Hasselt, J.G.C. Unraveling Antimicrobial Resistance Using Metabolomics. Drug Discov. Today 2022, 27, 1774–1783. [Google Scholar] [CrossRef] [PubMed]
- la Rosa, R.; Johansen, H.K.; Molina, S. Convergent Metabolic Specialization through Distinct Evolutionary Paths in Pseudomonas Aeruginosa. mBio 2018, 9, e00269-18. [Google Scholar] [CrossRef] [PubMed]
- Lanza, V.F.; Baquero, F.; Martinez, J.L.; Ramos-Ruiz, R.; González-Zorn, B.; Andremont, A.; Sánchez-Valenzuela, A.; Ehrlich, S.D.; Kennedy, S.; Ruppé, E.; et al. In-depth resistome analysis by targeted metagenomics. Microbiome 2018, 6, 11. [Google Scholar] [CrossRef]
- Petrova, O.E.; Schurr, J.R.; Schurr, M.J.; Sauer, K. Microcolony Formation by the Opportunistic Pathogen Pseudomonas Aeruginosa Requires Pyruvate Andpyruvate Fermentation. Mol. Microbiol. 2012, 86, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Schelli, K.; Zhong, F.; Zhu, J. Comparative Metabolomics Revealing Staphylococcus Aureus Metabolic Response to Different Antibiotics. Microb. Biotechnol. 2017, 10, 1764–1774. [Google Scholar] [CrossRef] [PubMed]
- Meylan, S.; Porter, C.B.M.; Yang, J.H.; Belenky, P.; Gutierrez, A.; Lobritz, M.A.; Park, J.; Kim, S.H.; Moskowitz, S.M.; Collins, J.J. Carbon Sources Tune Antibiotic Susceptibility in Pseudomonas Aeruginosa via Tricarboxylic Acid Cycle Control. Cell Chem. Biol. 2017, 24, 195–206. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, M.J.; Peng, B.; Peng, X.; Li, H. Reduced ROS-Mediated Antibiotic Resistance and Its Reverting by Glucose in Vibrio Alginolyticus. Environ. Microbiol. 2020, 22, 4367–4380. [Google Scholar] [CrossRef]
- Wang, C.; Dong, X.S.; Yang, Y.Y.; Xu, G.J.; Wu, M.M.; Yan, F.J.; Zhang, L.G.; An, L.; Fu, P.S.; Wang, X.R.; et al. Metabolites in the TCA Cycle Promote Resistance to Chloramphenicol of Edwardsiella tarda. J. Proteome Res. 2021, 20, 972–981. [Google Scholar] [CrossRef]
- Han, M.-L.; Zhu, Y.; Creek, D.J.; Lin, Y.-W.; Gutu, A.D.; Hertzog, P.; Purcell, T.; Shen, H.-H.; Moskowitz, S.M.; Velkov, T.; et al. Comparative Metabolomics and Transcriptomics Reveal Multiple Pathways Associated with Polymyxin Killing in Pseudomonas aeruginosa. mSystems 2019, 4, e00149-18. [Google Scholar] [CrossRef]
- Zampieri, M.; Zimmermann, M.; Claassen, M.; Sauer, U. Nontargeted Metabolomics Reveals the Multilevel Response to Antibiotic Perturbations. Cell Rep. 2017, 19, 1214–1228. [Google Scholar] [CrossRef]
- Edwards, J.S.; Palsson, B.O. Systems Properties of the Haemophilus Influenzae Rd Metabolic Genotype. J. Biol. Chem. 1999, 274, 17410–17416. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.S.; Palsson, B.O. The Escherichia coli MG1655 in Silico Metabolic Genotype: Its Definition, Characteristics, and Capabilities. Proc. Natl. Acad. Sci. USA 2000, 97, 5528–5533. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.J.; Monk, J.M.; Palsson, B.O. Using Genome-Scale Models to Predict Biological Capabilities. Cell 2015, 161, 971–987. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Palsson, B. A Protocol for Generating a High-Quality Genome-Scale Metabolic Reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef] [PubMed]
- Price, N.; Reed, J.; Palsson, B. Genome-scale models of microbial cells: Evaluating the consequences of constraints. Nat. Rev. Genet. 2004, 2, 886–897. [Google Scholar] [CrossRef]
- Piubeli, F.; Salvador, M.; Argandoña, M.; Nieto, J.J.; Bernal, V.; Pastor, J.M.; Cánovas, M.; Vargas, C. Insights into metabolic osmoadaptation of the ectoines-producer bacterium Chromohalobacter salexigens through a high-quality genome scale metabolic model. Microb. Cell Factories 2018, 17, 2. [Google Scholar] [CrossRef]
- Kim, T.Y.; Kim, H.U.; Lee, S.Y. Metabolite-Centric Approaches for the Discovery of Antibacterials Using Genome-Scale Metabolic Networks. Metab. Eng. 2010, 12, 105–111. [Google Scholar] [CrossRef]
- Jensen, P.A.; Zhu, Z.; van Opijnen, T. Antibiotics Disrupt Coordination between Transcriptional and Phenotypic Stress Responses in Pathogenic Bacteria. Cell Rep. 2017, 20, 1705–1716. [Google Scholar] [CrossRef]
- Zhu, Y.; Czauderna, T.; Zhao, J.; Klapperstueck, M.; Maifiah, M.H.M.; Han, M.L.; Lu, J.; Sommer, B.; Velkov, T.; Lithgow, T.; et al. Genome-Scale Metabolic Modeling of Responses to Polymyxins in Pseudomonas aeruginosa. Gigascience 2018, 7, giy021. [Google Scholar] [CrossRef]
- Banerjee, D.; Raghunathan, A. Constraints-Based Analysis Identifies NAD + Recycling through Metabolic Reprogramming in Antibiotic Resistant Chromobacterium violaceum. PLoS ONE 2019, 14, e0210008. [Google Scholar] [CrossRef]
- Dunphy, L.J.; A Papin, J. Biomedical applications of genome-scale metabolic network reconstructions of human pathogens. Curr. Opin. Biotechnol. 2017, 51, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Bartell, J.A.; Blazier, A.S.; Yen, P.; Thøgersen, J.C.; Jelsbak, L.; Goldberg, J.B.; Papin, J.A. Reconstruction of the metabolic network of Pseudomonas aeruginosa to interrogate virulence factor synthesis. Nat. Commun. 2017, 8, 14631. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Parmar, D.; Bhattacharya, N.; Ghanate, A.D.; Panchagnula, V.; Raghunathan, A. A Scalable Metabolite Supplementation Strategy against Antibiotic Resistant Pathogen Chromobacterium violaceum Induced by NAD+/NADH+ Imbalance. BMC Syst. Biol. 2017, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Christopher Noone, J.; Helmersen, K.; Leegaard, T.M.; Skråmm, I.; Aamot, H.V. Article Rapid Diagnostics of Orthopaedic-Implant-Associated Infections Using Nanopore Shotgun Metagenomic Sequencing on Tissue Biopsies. Microorganisms 2021, 9, 97. [Google Scholar] [CrossRef]
- Bernstein, D.B.; Sulheim, S.; Almaas, E.; Segrè, D. Addressing Uncertainty in Genome-Scale Metabolic Model Reconstruction and Analysis. Genome Biol. 2021, 22, 64. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tool | Reference | |
|---|---|---|
| ARG-ANNOT | [20] | |
| CARD/RGI | [21] | |
| ARGs | AMRFinder | [22] |
| ResFinder | [23] | |
| PointFinder | [24] | |
| Fastp | [25] | |
| Preprocessing and assembly-WGS | SPAdes/ | [26] |
| Flye | [27] | |
| Mzmine3 | [28] | |
| MetaboAnalyst 5.0 | [29] | |
| Metabolomic analysis | MetFlow | [30] |
| Omicsnet | [31] | |
| PaintOmics 3 | [32] | |
| DESeq | [33] | |
| edgeR | [34] | |
| General tools for transcriptomic analysis | limma | [35] |
| HTseq | [36] | |
| Rcount | [37] | |
| Cufflinks-Cuffdiff | [38] | |
| BioCyc | [39] | |
| BioMet ToolBox 2.0 | [40] | |
| GEM reconstruction | Kegg | [41] |
| GeneOntology | [42] | |
| Pathway-tools | [43] | |
| GEM analysis | CobraToolbox | [44] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francine, P. Systems Biology: New Insight into Antibiotic Resistance. Microorganisms 2022, 10, 2362. https://doi.org/10.3390/microorganisms10122362
Francine P. Systems Biology: New Insight into Antibiotic Resistance. Microorganisms. 2022; 10(12):2362. https://doi.org/10.3390/microorganisms10122362
Chicago/Turabian StyleFrancine, Piubeli. 2022. "Systems Biology: New Insight into Antibiotic Resistance" Microorganisms 10, no. 12: 2362. https://doi.org/10.3390/microorganisms10122362
APA StyleFrancine, P. (2022). Systems Biology: New Insight into Antibiotic Resistance. Microorganisms, 10(12), 2362. https://doi.org/10.3390/microorganisms10122362