
Characterization and Dynamics of the Gut Microbiota in Rice Fishes at Different Developmental Stages in Rice-Fish Coculture Systems


Abstract
:1. Introduction
2. Materials and Methods
2.1. Co-Culturing and Sample Collection
2.2. DNA Extraction and 16S rRNA Gene Sequencing
2.3. Bioinformatics and Amplicon Sequencing Data Analyses
3. Results
3.1. Overview of Sequencing Data
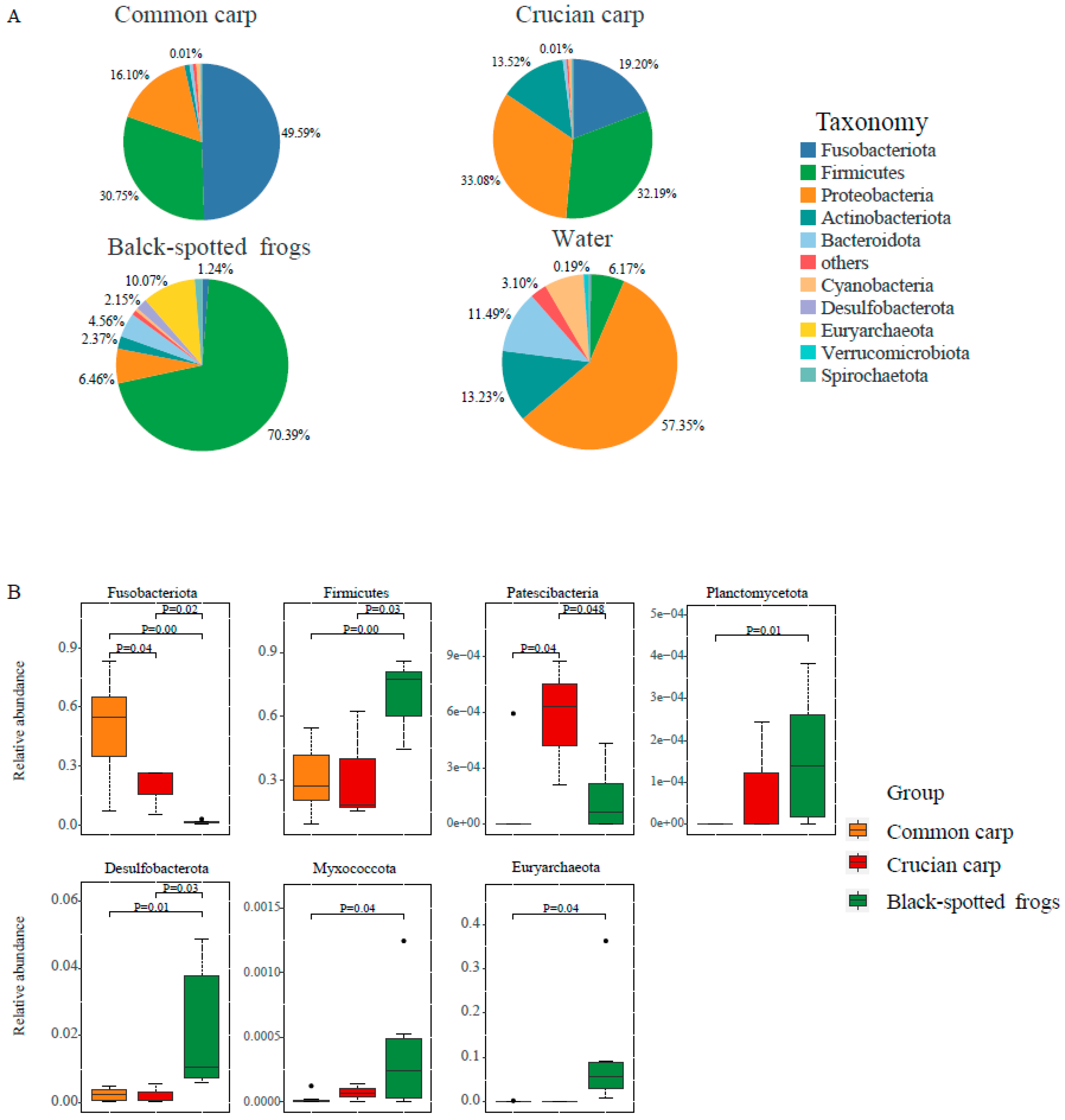
3.2. Gut Microbial Community of Three Sympatric Aquatic Animals
3.2.1. Characteristics of Microbial Composition in Three Sympatric Species
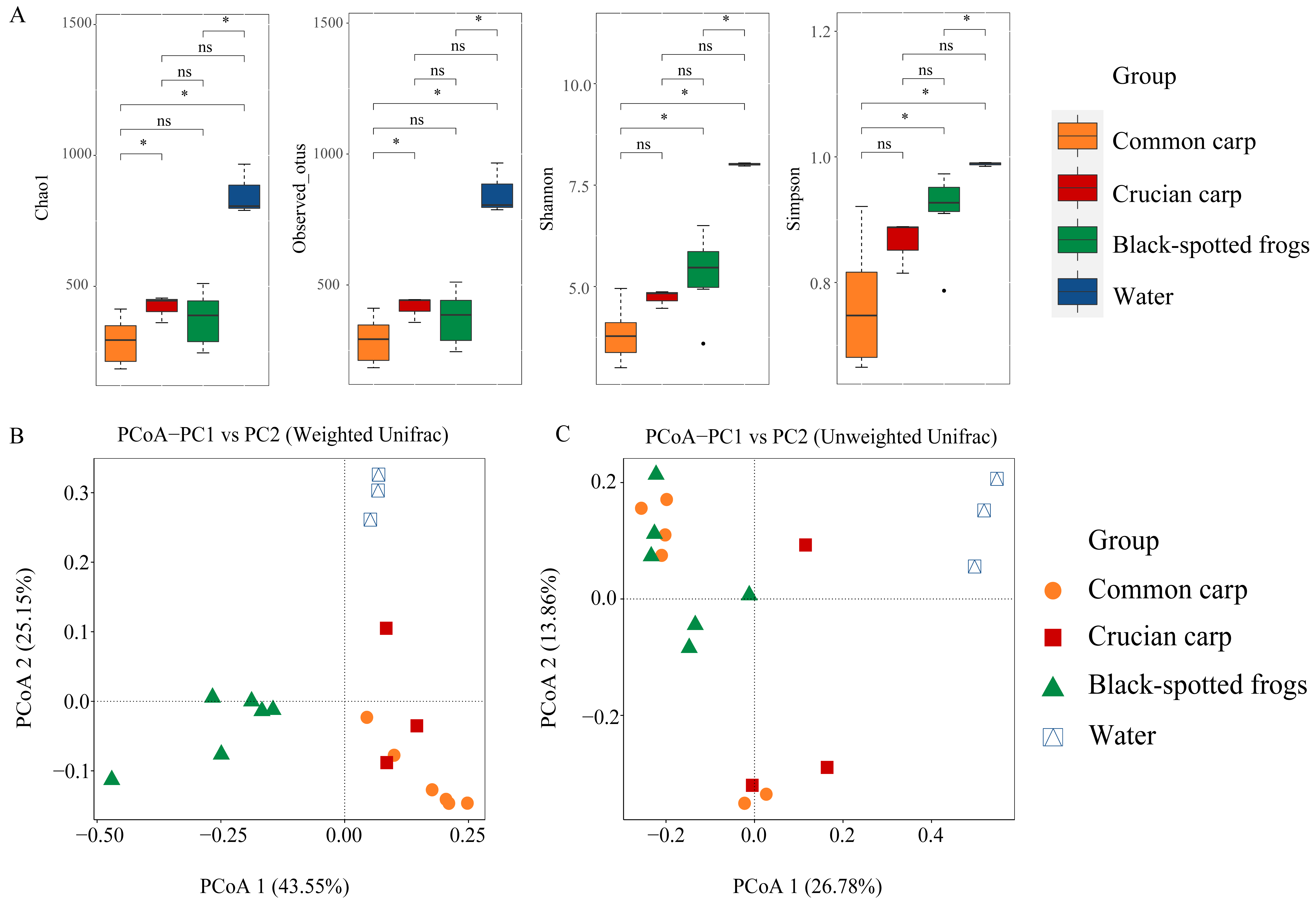
3.2.2. Alpha and Beta Diversity of Gut Microbiota among Three Sympatric Aquatic Species
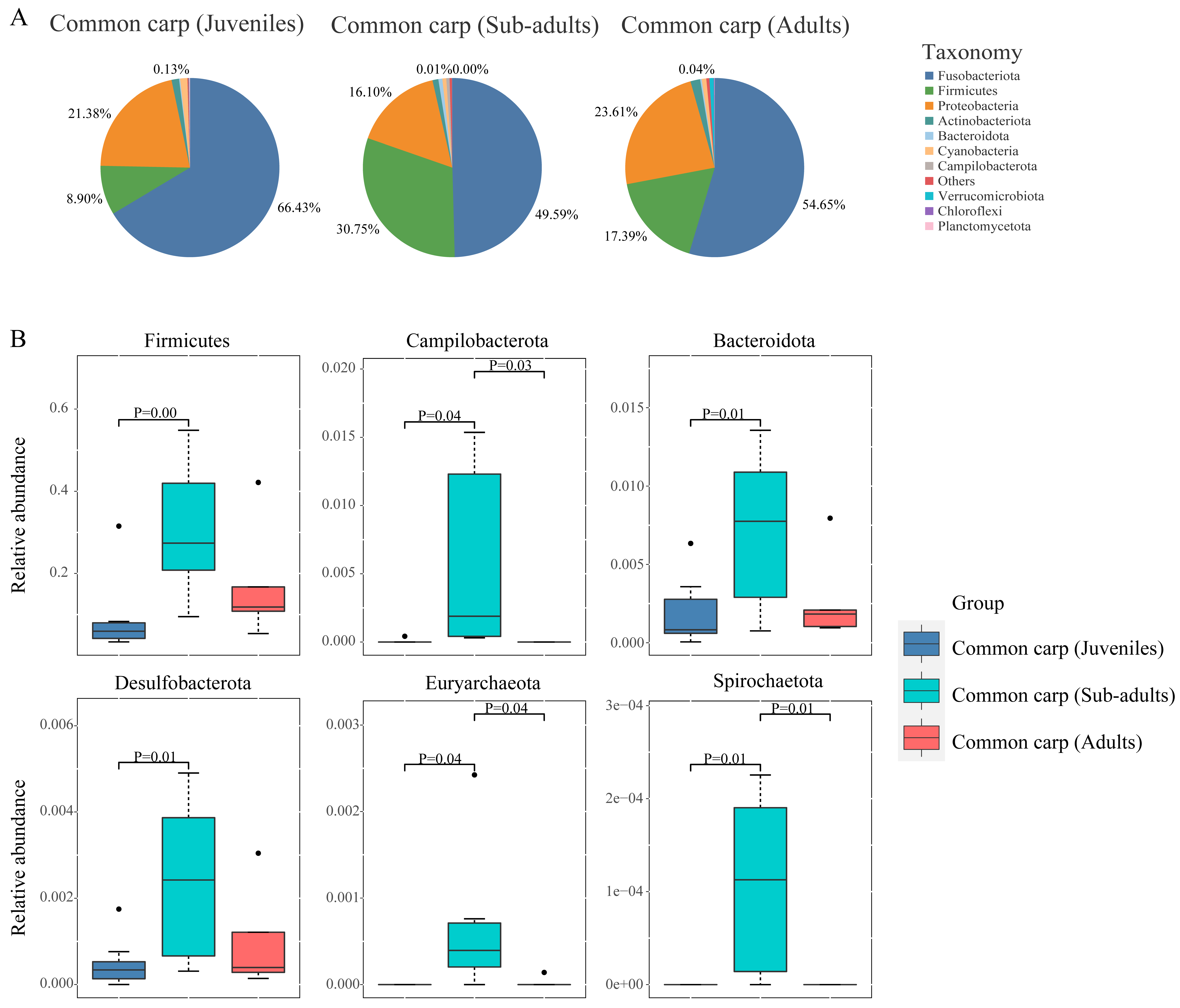
3.3. Temporal Variations in Gut Microbiota of Common Carp during Different Development Stages
3.3.1. Temporal Changes in Microbial Composition
3.3.2. Alpha and Beta Diversity of Gut Microbiota during Three Different Developmental Stages
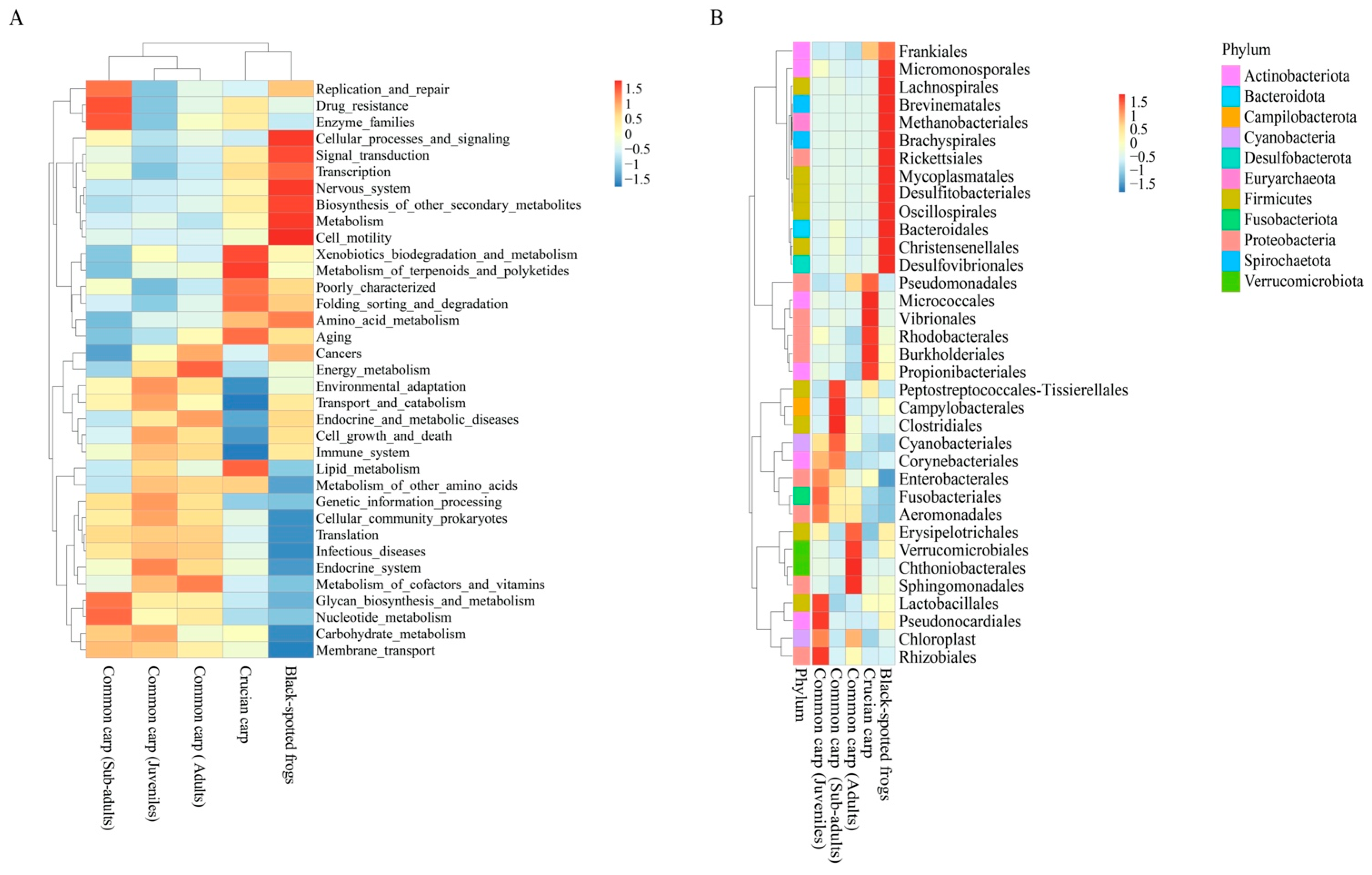
3.4. Functional Potential of Bacterial Community
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Xie, J.; Hu, L.L.; Tang, J.J.; Wu, X.; Li, N.N.; Yuan, Y.G.; Yang, H.S.; Zhang, J.; Luo, S.M.; Chen, X. Ecological mechanisms underlying the sustainability of the agricultural heritage rice-fish coculture system. Proc. Natl. Acad. Sci. USA 2011, 108, E1381–E1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frei, M.; Becker, K. Integrated rice–fish production and methane emission under greenhouse conditions. Agric. Ecosyst. Environ. 2005, 107, 51–56. [Google Scholar] [CrossRef]
- Ahmed, N.; Turchini, G.M. The evolution of the blue-green revolution of rice-fish cultivation for sustainable food production. Sustain. Sci. 2021, 16, 1375–1390. [Google Scholar]
- Hu, Y.J.; Cai, M.L.; Chu, W.Y.; Hu, Y. Dysbiosis of Gut Microbiota and Lipidomics of Content Induced by Dietary Methionine Restriction in Rice Field Eel (Monopterus albus). Front. Microbiol. 2022, 13, 917051. [Google Scholar] [CrossRef]
- Guo, L.; Zhao, L.F.; Ye, J.L.; Ji, Z.J.; Tang, J.J.; Bai, K.Y.; Zheng, S.J.; Hu, L.L.; Chen, X. Using aquatic animals as partners to increase yield and maintain soil nitrogen in the paddy ecosystems. eLife 2022, 11, e73869. [Google Scholar] [CrossRef]
- Ahmed, N.; Garnett, S.T. Integrated rice-fish farming in Bangladesh: Meeting the challenges of food security. Food Secur. 2011, 3, 81–92. [Google Scholar] [CrossRef]
- Ahmed, N.; Wahab, M.A.; Thilsted, S.H. Integrated aquaculture-agriculture systems in Bangladesh: Potential for sustainable livelihoods and nutritional security of the rural poor. Aquac. Asia 2007, 12, 14–22. [Google Scholar]
- Hu, L.L.; Ren, W.Z.; Tang, J.J.; Li, N.N.; Zhang, J.; Chen, X. The productivity of traditional rice–fish co-culture can be increased without increasing nitrogen loss to the environment. Agric. Ecosyst. Environ. 2013, 177, 28–34. [Google Scholar] [CrossRef]
- Hu, L.L.; Zhang, J.; Ren, W.Z.; Guo, L.; Cheng, Y.X.; Li, J.Y.; Li, K.X.; Zhu, Z.W.; Zhang, J.E.; Luo, S.M.; et al. Can the co-cultivation of rice and fish help sustain rice production? Sci. Rep. 2016, 6, 28728. [Google Scholar] [CrossRef] [Green Version]
- Dwiyana, E.; Mendoza, T. Determinants of productivity and profitability of rice-fish farming systems. Asia Life Sci. 2008, 17, 21–42. [Google Scholar]
- Zhang, J.; Hu, L.L.; Ren, W.Z.; Guo, L.; Tang, J.J.; Shu, M.A.; Chen, X. Rice-soft shell turtle coculture effects on yield and its environment. Agric. Ecosyst. Environ. 2016, 224, 116–122. [Google Scholar] [CrossRef]
- Yang, S.; Du, J.; Luo, J.; Zhou, Y.; Long, Y.; Xu, G.; Zhao, L.; Du, Z.; Yan, T. Effects of different diets on the intestinal microbiota and immunity of common carp (Cyprinus carpio). J. Appl. Microbiol. 2019, 127, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ni, J.; Li, J.; Wang, C.; Li, X.; Wu, S.; Zhang, T.; Yu, Y.; Yan, Q. Comparative study on gastrointestinal microbiota of eight fish species with different feeding habits. J. Appl. Microbiol. 2014, 117, 1750–1760. [Google Scholar] [CrossRef]
- Liu, H.; Guo, X.W.; Gooneratne, R.; Lai, R.F.; Zeng, C.; Zhan, F.B.; Wang, W.M. The gut microbiome and degradation enzyme activity of wild freshwater fishes influenced by their trophic levels. Sci. Rep. 2016, 6, 24340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, H. Pesticide use in rice and rice-fish farms in the Mekong Delta, Vietnam. Crop Prot. 2001, 20, 897–905. [Google Scholar] [CrossRef]
- Haroon, A.K.Y.; Pittman, K.A. Rice-fish culture: Feeding, growth and yield of two size classes of Puntius gonionotus Bleeker and Oreochromis spp. in Bangladesh. Aquaculture 1997, 154, 261–281. [Google Scholar] [CrossRef]
- Little, D.C.; Surintaraseree, P.; Innes-Taylor, N. Fish culture in rainfed rice fields of northeast Thailand. Aquaculture 1996, 140, 295–321. [Google Scholar] [CrossRef]
- Lu, J.B.; Li, X. Review of rice–fish-farming systems in China—One of the Globally Important Ingenious Agricultural Heritage Systems (GIAHS). Aquaculture 2006, 260, 106–113. [Google Scholar] [CrossRef]
- Peng, M.; Xue, J.J.; Hu, Y.; Wen, C.G.; Hu, B.Q.; Jian, S.Q.; Liang, L.F.; Yang, G. Disturbance in the homeostasis of intestinal microbiota by a high-fat diet in the rice field eel (Monopterus albus). Aquaculture 2019, 502, 347–355. [Google Scholar] [CrossRef]
- Meng, X.L.; Hu, W.P.; Wu, S.K.; Zhu, Z.X.; Lu, R.H.; Yang, G.K.; Qin, C.B.; Yang, L.P.; Nie, G.X. Chinese yam peel enhances the immunity of the common carp (Cyprinus carpio L.) by improving the gut defence barrier and modulating the intestinal microflora. Fish Shellfish. Immunol. 2019, 95, 528–537. [Google Scholar] [CrossRef]
- Yang, S.; Duan, Y.L.; Zhang, J.; Zhou, J.; Liu, Y.; Du, J.; Zhao, L.L.; Du, Z.J.; Han, S.S. Observational comparisons of intestinal microbiota characterizations, immune enzyme activities, and muscle amino acid compositions of loach in paddy fields and ponds in Sichuan Province. Appl. Microbiol. Biotechnol. 2017, 101, 4775–4789. [Google Scholar] [CrossRef]
- Zhu, H.J.; Qiang, J.; Xu, G.C.; Tao, Y.F.; Bao, J.W.; Xu, P. Microbial community structure of hybrid yellow catfish in rice-fish co-culture system in hani terrace. Acta Hydrobiol. Sin. 2021, 45, 1232–1242. (In Chinese) [Google Scholar]
- Dehler, C.E.; Secombes, C.J.; Martin, S.A.M. Environmental and physiological factors shape the gut microbiota of Atlantic salmon parr (Salmo salar L.). Aquaculture 2017, 467, 149–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanasiri, A.K.S.; Brunvold, L.; Brinchmann, M.F.; Korsnes, K.; Bergh, Ø.; Kiron, V. Changes in the Intestinal Microbiota of Wild Atlantic cod Gadus morhua L. Upon Captive Rearing. Microb. Ecol. 2011, 61, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Rawls, J.F.; Mahowald, M.A.; Ley, R.E.; Gordon, J.I. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell 2006, 127, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef]
- Clements, K.D.; Angert, E.R.; Montgomery, W.L.; Choat, J.H. Intestinal microbiota in fishes: What’s known and what’s not. Mol. Ecol. 2014, 23, 1891–1898. [Google Scholar] [CrossRef]
- Kamada, N.; Seo, S.U.; Chen, G.Y.; Núñez, G. Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 2013, 13, 321–335. [Google Scholar] [CrossRef]
- Wang, A.R.; Chao, R.; Ringø, E.; Zhou, Z.G. Progress in fish gastrointestinal microbiota research. Rev. Aquac. 2018, 10, 626–640. [Google Scholar] [CrossRef] [Green Version]
- Llewellyn, M.; Boutin, S.; Hoseinifar, S.H.; Derome, N. Teleost microbiomes: The state of the art in their characterization, manipulation and importance in aquaculture and fisheries. Front. Microbiol. 2014, 5, 207. [Google Scholar] [CrossRef] [Green Version]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogra, S.K.; Doré, J.; Damak, S. Gut Microbiota Resilience: Definition, Link to Health and Strategies for Intervention. Front. Microbiol. 2020, 11, 572921. [Google Scholar] [CrossRef] [PubMed]
- Li, X.H.; Zhou, L.; Yu, Y.H.; Ni, J.J.; Xu, W.J.; Yan, Q.Y. Composition of Gut Microbiota in the Gibel Carp (Carassius auratus gibelio) Varies with Host Development. Microb. Ecol. 2017, 74, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, P.; Fuentes, P.; De La Fuente, L.; Barros, L.; Magne, F.; Opazo, R.; Ibacache, C.; Espejo, R.; Romero, J. Short-term effects of dietary soybean meal and lactic acid bacteria on the intestinal morphology and microbiota of Atlantic salmon (Salmo salar). Aquac. Nutr. 2013, 19, 827–836. [Google Scholar] [CrossRef]
- Wong, S.; Waldrop, T.; Summerfelt, S.; Davidson, J.; Barrows, F.; Kenney, P.B.; Welch, T.; Wiens, G.D.; Snekvik, K.; Rawls, J.F.; et al. Aquacultured rainbow trout (Oncorhynchus mykiss) possess a large core intestinal microbiota that is resistant to variation in diet and rearing density. Appl. Environ. Microbiol. 2013, 79, 4974–4984. [Google Scholar] [CrossRef] [Green Version]
- Ingerslev, H.C.; Strube, M.L.; Jørgensen, L.V.; Dalsgaard, I.; Boye, M.; Madsen, L. Diet type dictates the gut microbiota and the immune response against Yersinia ruckeri in rainbow trout (Oncorhynchus mykiss). Fish Shellfish. Immunol. 2014, 40, 624–633. [Google Scholar] [CrossRef]
- Sullam, K.E.; Essinger, S.D.; Lozupone, C.A.; O’Connor, M.P.; Rosen, G.L.; Knight, R.; Kilham, S.S.; Russell, J.A. Environmental and ecological factors that shape the gut bacterial communities of fish: A meta-analysis. Mol. Ecol. 2012, 21, 3363–3378. [Google Scholar] [CrossRef] [Green Version]
- Givens, C.E.; Ransom, B.; Bano, N.; Hollibaugh, J.T. Comparison of the gut microbiomes of 12 bony fish and 3 shark species. Mar. Ecol. Prog. Ser. 2015, 518, 209–223. [Google Scholar] [CrossRef] [Green Version]
- Li, X.M.; Zhu, Y.J.; Yan, Q.Y.; Ringø, E.; Yang, D.G. Do the intestinal microbiotas differ between paddlefish (Polyodon spathala) and bighead carp (Aristichthys nobilis) reared in the same pond? J. Appl. Microbiol. 2015, 117, 1245–1252. [Google Scholar] [CrossRef]
- Nayak, S.K. Role of gastrointestinal microbiota in fish. Aquac. Res. 2010, 41, 1553–1573. [Google Scholar]
- Fraune, S.; Bosch, T. Why bacteria matter in animal development and evolution. BioEssays 2010, 32, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Q.Y.; van der Gast, C.J.; Yu, Y.H. Bacterial community assembly and turnover within the intestines of developing zebrafish. PLoS ONE 2012, 7, e30603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, W.Z.; Burns, A.R.; Stagaman, K.; Wong, S.; Rawls, J.F.; Guillemin, K.; Bohannan, B.J. The composition of the zebrafish intestinal microbial community varies across development. ISME J. 2016, 10, 644–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, F.; Zhu, W.; Yu, Y.; He, Z.; Wu, B.; Wang, C.; Shu, L.; Li, X.; Yin, H.; Wang, J.; et al. Host development overwhelms environmental dispersal in governing the ecological succession of zebrafish gut microbiota. NPJ Biofilms Microbiomes 2021, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Bakke, I.; Coward, E.; Andersen, T.; Vadstein, O. Selection in the host structures the microbiota associated with developing cod larvae (Gadus morhua). Environ. Microbiol. 2015, 17, 3914–3924. [Google Scholar] [CrossRef]
- Zhang, Z.M.; Li, D.P.; Refaey, M.M.; Xu, W.T.; Tang, R.; Li, L. Host Age Affects the Development of Southern Catfish Gut Bacterial Community Divergent from That in the Food and Rearing Water. Front. Microbiol. 2018, 9, 495. [Google Scholar] [CrossRef]
- Lokesh, J.; Kiron, V.; Sipkema, D.; Fernandes, J.M.O.; Moum, T. Succession of embryonic and the intestinal bacterial communities of Atlantic salmon (Salmo salar) reveals stage-specific microbial signatures. MicrobiologyOpen 2019, 8, e00672. [Google Scholar] [CrossRef]
- Bledsoe, J.W.; Peterson, B.C.; Swanson, K.S.; Small, B.C. Ontogenetic Characterization of the Intestinal Microbiota of Channel Catfish through 16S rRNA Gene Sequencing Reveals Insights on Temporal Shifts and the Influence of Environmental Microbes. PLoS ONE 2016, 11, e0166379. [Google Scholar] [CrossRef] [Green Version]
- Rosado, D.; Pérez-Losada, M.; Pereira, A.; Severino, R.; Xavier, R. Effects of aging on the skin and gill microbiota of farmed seabass and seabream. Anim. Microbiome 2021, 3, 10. [Google Scholar] [CrossRef]
- Shen, X.D.; Gou, W.M. Research on rice paddy development and propects in china. Chin. Fish. Econ. 2013, 31, 151–156. [Google Scholar]
- Li, T.; Long, M.; Gatesoupe, F.J.; Zhang, Q.Q.; Li, A.H.; Gong, X.N. Comparative Analysis of the Intestinal Bacterial Communities in Different Species of Carp by Pyrosequencing. Microb. Ecol. 2015, 69, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Rodríguez, M.; Scheifler, M.; Sanchez-Brosseau, S.; Magnanou, E.; West, N.; Suzuki, M.; Duperron, S.; Desdevises, Y. Host Species and Body Site Explain the Variation in the Microbiota Associated to Wild Sympatric Mediterranean Teleost Fishes. Microb. Ecol. 2020, 80, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.Y.; Wang, Z.W.; Wan, Q.H.; Fang, S.G. Metagenomics Reveals Seasonal Functional Adaptation of the Gut Microbiome to Host Feeding and Fasting in the Chinese Alligator. Front. Microbiol. 2019, 10, 2409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K. QIIME allows integration and analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Wilcoxon, F. Individual Comparisons by Ranking Methods. Biometrics 1945, 1, 196–202. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; O’Hara, R.G.; Simpson, G.L.; Solymos, P.; Stevens, H.; Wagner, H.W. Multivariate analysis of ecological communities in R: Vegan tutorial. R package version 1.7. 2013. pp. 1–43.
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.R.; Nagarajan, N.; Pop, M. Statistical Methods for Detecting Differentially Abundant Features in Clinical Metagenomic Samples. PLoS Comput. Biol. 2009, 5, e1000352. [Google Scholar] [CrossRef] [PubMed]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, Z.J.; Xu, G.C.; Shao, N.L.; Wang, B.Z.; Gao, J.C.; Xu, P.; He, J. Comparison of gut microbiota in carps from fish monoculture ponds and the rice-fish co-culture system in Hani Terraces. Acta Microbiol. Sin. 2022, 62, 1473–1484. (In Chinese) [Google Scholar]
- Hu, Y.J.; Zhang, J.Z.; Xue, J.J.; Chu, W.Y.; Hu, Y. Effects of Dietary Soy Isoflavone and Soy Saponin on Growth Performance, Intestinal Structure, Intestinal Immunity and Gut Microbiota Community on Rice Field Eel (Monopterus albus). Aquaculture 2020, 537, 736506. [Google Scholar] [CrossRef]
- Li, J.J.; Haffner, G.D.; Wang, D.Y.; Zhang, L.; Li, Y.; Deng, H.T.; Drouillard, K.G. Protein and lipid growth rates regulate bioaccumulation of PCBs and Hg in Bighead Carp (Hypophthalmichthys nobilis) and Silver Carp (Hypophthalmichthys molitrix) from the Three Gorges Reservoir, China. Environ. Pollut. 2018, 243, 152–162. [Google Scholar] [CrossRef]
- Shu, Y.L.; Hong, P.; Yu, Q.; Wang, G.; Zhang, J.H.; Donde, O.O.; Xiao, B.D.; Wu, H.L. High-Throughput Sequencing Analysis Reveals Correlations between Host Phylogeny, Gut Microbiota, and Habitat of Wild Frogs from a Mountainous Area. Copeia 2019, 107, 131–137. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, J.Q.; Liu, Y.; Zhang, J.; Chen, X.H.; Qu, Y.F. Comparative study on gut microbiota in three Anura frogs from a mountain stream. Ecol. Evol. 2022, 12, e8854. [Google Scholar] [CrossRef]
- Corrigan, A.; de Leeuw, M.; Penaud-Frézet, S.; Dimova, D.; Murphy, R.A. Phylogenetic and functional alterations in bacterial community compositions in broiler ceca as a result of mannan oligosaccharide supplementation. Appl. Environ. Microbiol. 2015, 81, 3460–3470. [Google Scholar] [CrossRef] [Green Version]
- Lopetuso, L.R.; Scaldaferri, F.; Petito, V.; Gasbarrini, A. Commensal Clostridia: Leading players in the maintenance of gut homeostasis. Gut Pathog. 2013, 5, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, Q.; Liu, X.N.; Hu, Z.F.; Ding, J.F.; Bie, J.; Wang, H.B.; Zhang, J.T. Effects of Captivity and Season on the Gut Microbiota of the Brown Frog (Rana dybowskii). Front. Microbiol. 2019, 10, 1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shortt, C.; Hasselwander, O.; Meynier, A.; Nauta, A.; Fernández, E.N.; Putz, P.; Rowland, I.; Swann, J.; Türk, J.; Vermeiren, J.; et al. Systematic review of the effects of the intestinal microbiota on selected nutrients and non-nutrients. Eur. J. Nutr. 2018, 57, 25–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, N.L.; Steven, B.; Penn, K.; Methé, B.A.; Detrich, W.H. Characterization of the intestinal microbiota of two Antarctic notothenioid fish species. Extremophiles 2009, 13, 679–685. [Google Scholar] [CrossRef]
- Ni, J.; Yan, Q.; Yu, Y.; Zhang, T. Factors influencing the grass carp gut microbiome and its effect on metabolism. FEMS Microbiol. Ecol. 2014, 87, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Ingerslev, H.C.; von Gersdorff Jørgensen, L.; Lenz Strube, M.; Larsen, N.; Dalsgaard, I.; Boye, M.; Madsen, L. The development of the gut microbiota in rainbow trout (Oncorhynchus mykiss) is affected by first feeding and diet type. Aquaculture 2014, 424–425, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Larsen, A.M. Studies on the Microbiota of Fishes and the Factors Influencing Their Composition. Doctoral Dissertation, Auburn University, Auburn, AL, USA, 2014. [Google Scholar]
- Hao, Y.T.; Wu, S.G.; Jakovli, I.; Zou, H.; Li, W.X.; Wang, G.T. Impacts of diet on hindgut microbiota and short-chain fatty acids in grass carp (Ctenopharyngodon idellus). Aquac. Res. 2017, 48, 5595–5605. [Google Scholar] [CrossRef]
- Nelson, T.M.; Rogers, T.L.; Brown, M.V. The Gut Bacterial Community of Mammals from Marine and Terrestrial Habitats. PLoS ONE 2013, 8, e83655. [Google Scholar] [CrossRef]
- Potrykus, J.; White, R.L.; Bearne, S.L. Proteomic investigation of amino acid catabolism in the indigenous gut anaerobe Fusobacterium varium. Proteomics 2008, 8, 2691–2703. [Google Scholar] [CrossRef]
- Zhou, Y.F.; Li, D.J.; Liu, D.Y.; Liu, J.B.; Yao, S.H.; Cai, Y.M. The Intestinal Flora Analysis of Four Types of Common Freshwater Fish. China Anim. Husb. Vet. Med. 2012, 39, 220–223. [Google Scholar]
- Moon, C.D.; Young, W.; Maclean, P.H.; Cookson, A.L.; Bermingham, E.N. Metagenomic insights into the roles of Proteobacteria in the gastrointestinal microbiomes of healthy dogs and cats. Microbiologyopen 2018, 7, e00677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giatsis, C.; Sipkema, D.; Smidt, H.; Heilig, H.; Benvenuti, G.; Verreth, J.; Verdegem, M. The impact of rearing environment on the development of gut microbiota in tilapia larvae. Sci. Rep. 2015, 5, 18206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevellec, M.; De Rome, N.; Bernatchez, L. Holobionts and ecological speciation: The intestinal microbiota of lake whitefish species pairs. Microbiome 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, T.X.; He, A.Y.; Lin, Y.F.; Huang, X.D.; Liu, L.; Zhou, L. Comparative analysis of microbial communities associated with the gill, gut, and habitat of two filter-feeding fish. Aquac. Rep. 2020, 18, 100501. [Google Scholar] [CrossRef]
- Sun, F.; Wang, C.; Chen, L.; Weng, G.; Zheng, Z. The intestinal bacterial community of healthy and diseased animals and its association with the aquaculture environment. Appl. Microbiol. Biotechnol. 2020, 104, 775–783. [Google Scholar] [CrossRef]
- Sehnal, L.; Brammer-Robbins, E.; Wormington, A.M.; Blaha, L.; Bisesi, J.; Larkin, I.; Martyniuk, C.J.; Simonin, M.; Adamovsky, O. Microbiome Composition and Function in Aquatic Vertebrates: Small Organisms Making Big Impacts on Aquatic Animal Health. Front. Microbiol. 2021, 12, 567408. [Google Scholar] [CrossRef]
- Li, T.; Li, H.; Gatesoupe, F.J.; She, R.; Lin, Q.; Yan, X.; Li, J.; Li, X. Bacterial Signatures of “Red-Operculum” Disease in the Gut of Crucian Carp (Carassius auratus). Microb. Ecol. 2017, 74, 510–521. [Google Scholar] [CrossRef]
- Li, J.; Gang, L.; Li, C.W.; Deng, Y.L.; Tadda, M.A.; Lan, L.H.; Zhu, S.M.; Liu, D.Z. Effects of different solid carbon sources on water quality, biofloc quality and gut microbiota of Nile tilapia (Oreochromis niloticus) larvae. Aquaculture 2018, 495, 919–931. [Google Scholar] [CrossRef]
- Zhang, X.Z.; You, Y.; Peng, F.; Tang, X.M.; Zhou, Y.F.; Liu, J.Y.; Lin, D.Q.; Zhou, Y.F. Interaction of Microbiota between Fish and the Environment of an In-Pond Raceway System in a Lake. Microorganisms 2022, 10, 1143. [Google Scholar] [CrossRef]
- Li, X.H.; Yu, Y.H.; Li, C.; Yan, Q.Y. Comparative study on the gut microbiotas of four economically important Asian carp species. Science China. Life Sci. 2018, 61, 696–705. [Google Scholar] [CrossRef]
- Wong, S.D.; Rawls, J.F. Intestinal microbiota composition in fishes is influenced by host ecology and environment. Mol. Ecol. 2012, 21, 3100–3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.; Stephens, W.Z.; Burns, A.R.; Stagaman, K.; David, L.A.; Bohannan, B.J.; Guillemin, K.; Rawls, J.F. Ontogenetic Differences in Dietary Fat Influence Microbiota Assembly in the Zebrafish Gut. mBio 2015, 6, e00687-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, J.; Navarrete, P. 16S rDNA-Based Analysis of Dominant Bacterial Populations Associated with Early Life Stages of Coho Salmon (Oncorhynchus kisutch). Microb. Ecol. 2006, 51, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, P.; Magne, F.; Araneda, C.; Fuentes, P.; Barros, L.; Opazo, R.; Espejo, R.; Romero, J. PCR-TTGE analysis of 16S rRNA from rainbow trout (Oncorhynchus mykiss) gut microbiota reveals host-specific communities of active bacteria. PLoS ONE 2012, 7, e31335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Yu, Y.; Feng, W.; Yan, Q.; Gong, Y. Host species as a strong determinant of the intestinal microbiota of fish larvae. J. Microbiol. 2012, 50, 29–37. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Flint, H.J.; Bayer, E.A.; Rincon, M.T.; Lamed, R.; White, B.A. Polysaccharide utilization by gut bacteria: Potential for new insights from genomic analysis. Nat. Rev. Microbiol. 2008, 6, 121–131. [Google Scholar] [CrossRef]
- Smriga, S.; Sandin, S.A.; Azam, F. Abundance, diversity, and activity of microbial assemblages associated with coral reef fish guts and feces. FEMS Microbiol. Ecol. 2010, 73, 31–42. [Google Scholar] [CrossRef]
- Finegold, S.M.; Vaisanen, M.L.; Molitoris, D.R.; Tomzynski, T.J.; Song, Y.; Liu, C.; Collins, M.D.; Lawson, P.A. Cetobacterium somerae sp. nov. from human feces and emended description of the genus Cetobacterium. Syst. Appl. Microbiol. 2003, 26, 177–181. [Google Scholar] [CrossRef]
- Kim, P.S.; Shin, N.-R.; Lee, J.-B.; Kim, M.-S.; Whon, T.W.; Hyun, D.-W.; Yun, J.-H.; Jung, M.-J.; Kim, J.Y.; Bae, J.-W. Host habitat is the major determinant of the gut microbiome of fish. Microbiome 2021, 9, 166. [Google Scholar] [CrossRef]
- Xiong, J.B.; Nie, L.; Chen, J. Current understanding on the roles of gut microbiota in fish disease and immunity. Zool. Res. 2019, 40, 70–76. [Google Scholar]
- Gerritsen, J.; Hornung, B.; Ritari, J.; Paulin, L.; Rijkers, G.T.; Schaap, P.J.; De Vos, W.M.; Smidt, H. A comparative and functional genomics analysis of the genus Romboutsia provides insight into adaptation to an intestinal lifestyle. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Gerritsen, J.; Hornung, B.; Renckens, B.; van Hijum, S.; Martins Dos Santos, V.A.P.; Rijkers, G.T.; Schaap, P.J.; de Vos, W.M.; Smidt, H. Genomic and functional analysis of Romboutsia ilealis CRIB(T) reveals adaptation to the small intestine. PeerJ 2017, 5, e3698. [Google Scholar] [CrossRef] [Green Version]
- Gerritsen, J.; Umanets, A.; Staneva, I.; Hornung, B.; Ritari, J.; Paulin, L.; Rijkers, G.T.; de Vos, W.M.; Smidt, H. Romboutsia hominis sp. nov., the first human gut-derived representative of the genus Romboutsia, isolated from ileostoma effluent. Int. J. Syst. Evol. Microbiol. 2018, 68, 3479–3486. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.T.; Roesch, L.F.W.; Ördberg, M.; Ilonen, J.; Atkinson, M.A.; Schatz, D.A.; Triplett, E.W.; Ludvigsson, J. Genetic risk for autoimmunity is associated with distinct changes in the human gut microbiome. Nat. Commun. 2019, 10, 3621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Fu, J.; Xiao, X.; Wang, F.; Jin, M.; Fang, W.; Wang, Y.; Zong, X. Faecal microbiota transplantation-mediated jejunal microbiota changes halt high-fat diet-induced obesity in mice via retarding intestinal fat absorption. Microb. Biotechnol. 2022, 15, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Muduli, C.; Tripathi, G.; Paniprasad, K.; Kumar, K.; Singh, R.K.; Rathore, G.J.B. Virulence potential of Aeromonas hydrophila isolated from apparently healthy freshwater food fish. Biologia 2021, 76, 1005–1015. [Google Scholar] [CrossRef]
- Mohammed, H.H.; Arias, C.R. Potassium permanganate elicits a shift of the external fish microbiome and increases host susceptibility to columnaris disease. Vet. Res. 2015, 46, 82. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pair-Wised Comparison | Weighted Unifrac | Unweighted Unifrac | ||
|---|---|---|---|---|
| R Value | p Value | R Value | p Value | |
| Common carp—Crucian carp | 0.284 | 0.159 | 0.617 | 0.015 |
| Common carp—Black-spotted frogs | 0.737 | 0.005 | 0.372 | 0.005 |
| Crucian carp—Black-spotted frogs | 0.574 | 0.005 | 0.648 | 0.015 |
| Black-spotted frogs—Water | 0.679 | 0.010 | 1 | 0.030 |
| Crucian carp—Water | 1 | 0.109 | 1 | 0.075 |
| Common carp—Water | 0.988 | 0.020 | 1 | 0.025 |
| Common carp (Juvenile)—Common carp (Sub-adult) | 0.318 | 0.035 | 0.442 | 0.005 |
| Common carp (Juvenile)—Common carp (Adult) | 0.213 | 0.085 | 0.204 | 0.075 |
| Common carp (Sub-adult)—Common carp (Adult) | 0.072 | 0.184 | 0.283 | 0.020 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tao, L.; Chai, J.; Liu, H.; Huang, W.; Zou, Y.; Wu, M.; Peng, B.; Wang, Q.; Tang, K. Characterization and Dynamics of the Gut Microbiota in Rice Fishes at Different Developmental Stages in Rice-Fish Coculture Systems. Microorganisms 2022, 10, 2373. https://doi.org/10.3390/microorganisms10122373
Tao L, Chai J, Liu H, Huang W, Zou Y, Wu M, Peng B, Wang Q, Tang K. Characterization and Dynamics of the Gut Microbiota in Rice Fishes at Different Developmental Stages in Rice-Fish Coculture Systems. Microorganisms. 2022; 10(12):2373. https://doi.org/10.3390/microorganisms10122373
Chicago/Turabian StyleTao, Ling, Jie Chai, Hongyi Liu, Wenhao Huang, Yan Zou, Mengling Wu, Buqing Peng, Qiong Wang, and Keyi Tang. 2022. "Characterization and Dynamics of the Gut Microbiota in Rice Fishes at Different Developmental Stages in Rice-Fish Coculture Systems" Microorganisms 10, no. 12: 2373. https://doi.org/10.3390/microorganisms10122373
APA StyleTao, L., Chai, J., Liu, H., Huang, W., Zou, Y., Wu, M., Peng, B., Wang, Q., & Tang, K. (2022). Characterization and Dynamics of the Gut Microbiota in Rice Fishes at Different Developmental Stages in Rice-Fish Coculture Systems. Microorganisms, 10(12), 2373. https://doi.org/10.3390/microorganisms10122373