
Exploring Diversity and Polymer Degrading Potential of Epiphytic Bacteria Isolated from Marine Macroalgae

 ,
, 
Abstract
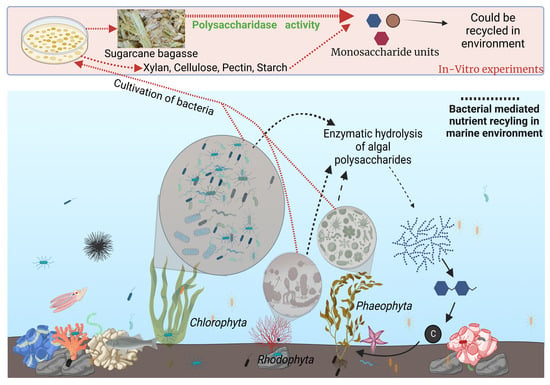
1. Introduction
2. Materials and Methods
2.1. Collection and Identification of Samples
2.2. Cultivation of Epiphytic Bacteria
2.3. Molecular Characterization of Bacterial Strains
2.4. Polymer Hydrolysis and Raw Substrate Degradation
3. Results and Discussion
3.1. Isolation, Identification and Phylogenetic Analysis
3.2. Analysis of Polymer Hydrolysis Potential
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roy, S.; Salvi, H.; Brahmbhatt, B.; Vaghela, N.; Das, L.; Pathak, B. Diversity and Distribution of Seaweeds in Selected Reefs and Island in Gulf of Kachchh. Seaweed Res. Utiln. 2015, 37, 12–19. [Google Scholar]
- Tait, L.W.; Schiel, D.R. Dynamics of productivity in naturally structured macroalgal assemblages: Importance of canopy structure on light-use efficiency. Mar. Ecol. Prog. Ser. 2011, 421, 97–107. [Google Scholar] [CrossRef][Green Version]
- Lachnit, T.; Blümel, M.; Imhoff, J.F.; Wahl, M. Specific epibacterial communities on macroalgae: Phylogeny matters more than habitat. Aquat. Biol. 2009, 5, 181–186. [Google Scholar] [CrossRef]
- Lachnit, T.; Meske, D.; Wahl, M.; Harder, T.; Schmitz, R. Epibacterial community patterns on marine macroalgae are host-specific but temporally variable. Environ. Microbiol. 2010, 13, 655–665. [Google Scholar] [CrossRef]
- Goecke, F.; Labes, A.; Wiese, J.; Imhoff, J.F. Chemical interactions between marine macroalgae and bacteria. Mar. Ecol. Prog. Ser. 2010, 409, 267–299. [Google Scholar] [CrossRef]
- Burke, C.; Thomas, T.; Lewis, M.; Steinberg, P.; Kjelleberg, S. Composition, uniqueness and variability of the epiphytic bacterial community of the green alga Ulva australis. ISME J. 2010, 5, 590–600. [Google Scholar] [CrossRef]
- Burke, C.; Steinberg, P.; Rusch, D.; Kjelleberg, S.; Thomas, T. Bacterial community assembly based on functional genes rather than species. Proc. Natl. Acad. Sci. USA 2011, 108, 14288–14293. [Google Scholar] [CrossRef]
- Leiva, S.; Alvarado, P.; Huang, Y.; Wang, J.; Garrido, I. Diversity of pigmented Gram-positive bacteria associated with marine macroalgae from Antarctica. FEMS Microbiol. Lett. 2015, 362. [Google Scholar] [CrossRef]
- Albakosh, M.A.; Naidoo, R.K.; Kirby, B.; Bauer, R. Identification of epiphytic bacterial communities associated with the brown alga Splachnidium rugosum. J. Appl. Phycol. 2015, 28, 1891–1901. [Google Scholar] [CrossRef]
- Martin, M.; Portetelle, D.; Michel, G.; Vandenbol, M. Microorganisms living on macroalgae: Diversity, interactions, and biotechnological applications. Appl. Microbiol. Biotechnol. 2014, 98, 2917–2935. [Google Scholar] [CrossRef]
- Potin, P.; Bouarab, K.; Salaün, J.; Pohnert, G.; Kloareg, B. Biotic interactions of marine algae. Curr. Opin. Plant Biol. 2002, 5, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Pasmore, M.; Costerton, J.W. Biofilms, bacterial signaling, and their ties to marine biology. J. Ind. Microbiol. Biotechnol. 2003, 30, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Florez, J.Z.; Camus, C.; Hengst, M.B.; Buschmann, A.H. A Functional Perspective Analysis of Macroalgae and Epiphytic Bacterial Community Interaction. Front. Microbiol. 2017, 8, 2561. [Google Scholar] [CrossRef]
- Selvarajan, R.; Sibanda, T.; Venkatachalam, S.; Ogola, H.J.O.; Obieze, C.C.; Msagati, T. Distribution, Interaction and Functional Profiles of Epiphytic Bacterial Communities from the Rocky Intertidal Seaweeds, South Africa. Sci. Rep. 2019, 9, 19835. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, N.S.; Naim, M.A.; Mohd-Nor, N.; Abidin, Z.A.Z. Diversity of cultivable bacteria by strategic enrichment isolated from farmed edible red seaweed, Gracilaria sp. J. CleanWAS 2020, 4, 17–20. [Google Scholar] [CrossRef]
- Hinojosa, V.S.; Asenjo, J.; Leiva, S. Agarolytic culturable bacteria associated with three antarctic subtidal macroalgae. World J. Microbiol. Biotechnol. 2018, 34, 73. [Google Scholar] [CrossRef]
- Naik, M.M.; Naik, D.; Charya, L.; Mujawar, S.Y.; Vaingankar, D.C. Application of Marine Bacteria Associated with Seaweed, Ulva lactuca, for Degradation of Algal Waste. Proc. Natl. Acad. Sci. India Sect. B Boil. Sci. 2018, 89, 1153–1160. [Google Scholar] [CrossRef]
- Odaneth, A.A. Diversity of Ulvan and Cellulose Depolymerizing Bacteria Associated with the Green Macroalgae ulva Spp. J. Appl. Biotechnol. Bioeng. 2017, 2, 136–142. [Google Scholar] [CrossRef][Green Version]
- Pappu, A.; Saxena, M.; Asolekar, S.R. Solid wastes generation in India and their recycling potential in building materials. Build. Environ. 2007, 42, 2311–2320. [Google Scholar] [CrossRef]
- Bernard, S.; Kazmin, A. Dirty Air: How India Became the Most Polluted Country on Earth. Financ. Times 2018, 12. [Google Scholar]
- Iqbal, N.; Agrawal, A.; Dubey, S.; Kumar, J. Role of Decomposers in Agricultural Waste Management. In Biotechnological Applications of Biomass; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef]
- Kumar, A.; Chandra, R. Ligninolytic enzymes and its mechanisms for degradation of lignocellulosic waste in environment. Heliyon 2020, 6, e03170. [Google Scholar] [CrossRef] [PubMed]
- Nayak, S.; Mukherjee, A.K. Management of Agricultural Wastes Using Microbial Agents. Waste Manag. Chall. Threat. Oppor. 2015, 65–91. [Google Scholar]
- Santanna, L.; Maria, M.; Pereira, N.; Bitancur, G.; Jaime, V.; Bevilaqua, J. Process for the Fermentative Production of Ethanol by Pichia Stipitis from the Hemicellulose Hydrolysate of Sugarcane Bagasse. WO Patent WO 2009/004273 Al, 8 January 2009. [Google Scholar]
- Rarbach, M.; Dragovic, Z. Efficient Lignocellulose Hydrolysis with Integrated Enzyme Production. EP Patent EP 2 471 940 A1, 2007. [Google Scholar]
- Dottori, F.A.; Ashley, R.; Benson, C.; Ca, B.; Benech, R. Bagasse Fractionation for Cellulosc Ethanoland Chemical Production. U.S. Patent 9,187,862 B2, 17 November 2015. [Google Scholar]
- Delmas, M.; Benjelloum, B.; Mlayah, B.B. Process for Producing Bioethanol by Enzymatic Hydrolysis of Cellulose. U.S. Patent 9,518,274B2, 13 December 2016. [Google Scholar] [CrossRef]
- Sakdaronnarong, C.; Jonglertjunya, W. Rice straw and sugarcane bagasse degradation mimicking lignocellulose decay in nature: An alternative approach to biorefinery. ScienceAsia 2012, 38, 364–372. [Google Scholar] [CrossRef]
- Bian, J.; Peng, P.; Peng, F.; Xiao, X.; Xu, F.; Sun, R.-C. Microwave-assisted acid hydrolysis to produce xylooligosaccharides from sugarcane bagasse hemicelluloses. Food Chem. 2014, 156, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Maeda, R.N.; Serpa, V.I.; Rocha, V.A.L.; Mesquita, R.A.A.; De Santa Anna, L.M.M.S.; Castro, A.M.; Driemeier, C.E.; Pereira, N.; Polikarpov, I. Enzymatic hydrolysis of pretreated sugar cane bagasse using Penicillium funiculosum and Trichoderma harzianum cellulases. Process. Biochem. 2011, 46, 1196–1201. [Google Scholar] [CrossRef]
- Doyle, J.; Doyle, J. A Rapid DNA Isolation Procedure for Small Quantities of Fresh Leaf Tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Saunders, G.W.; McDevit, D.C. Methods for DNA Barcoding Photosynthetic Protists Emphasizing the Macroalgae and Diatoms. Methods Mol. Biol. 2012, 858, 207–222. [Google Scholar] [CrossRef]
- Verma, A.; Sundharam, S.S.; Pal, Y.; Bisht, B.; Yadav, P.; Krishnamurthi, S. Yangia mangrovi sp. nov., a novel member of the Roseobacter clade isolated from mangrove soil and emended description of Yangia pacifica Dai et al. 2006. Int. J. Syst. Evol. Microbiol. 2021, 71, 005021. [Google Scholar] [CrossRef]
- Sahoo, D.; Debasish, N. Seaweeds of Indian Coast; APH Pub. Corp.: Delhi, India, 2001. [Google Scholar]
- Dhargalkar, V.; Aquaculture, X.V. Undefined Southern Ocean Seaweeds: A Resource for Exploration in Food and Drugs; Elsevier: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Pitcher, D.G.; Saunders, N.A.; Owen, R.J. Rapid extraction of bacterial genomic DNA with guanidium thiocyanate. Lett. Appl. Microbiol. 1989, 8, 151–156. [Google Scholar] [CrossRef]
- Ojha, A.K.; Verma, A.; Pal, Y.; Bhatt, D.; Mayilraj, S.; Krishnamurthi, S. Marinomonas epiphytica sp. nov., isolated from a marine intertidal macroalga. Int. J. Syst. Evol. Microbiol. 2017, 67, 2746–2751. [Google Scholar] [CrossRef]
- Yoon, S.-H.; Ha, S.-M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, C.W.; Doyle, J.D.; Hugley, B. A new solid medium for enumerating cellulose-utilizing bacteria in soil. Appl. Environ. Microbiol. 1995, 61, 2016–2019. [Google Scholar] [CrossRef]
- Catherine, G.V. Isolation of Cellulose Degrading Bacteria and Yeasts From Pineapple Waste. Int. J. Curr. Res. Rev. 2012, 4, 7. [Google Scholar]
- Awedem Wobiwo, F.; Chaturvedi, T.; Boda, M.; Fokou, E.; Emaga, T.H.; Cybulska, I.; Deleu, M.; Gerin, P.A.; Thomsen, M.H. Bioethanol Potential of Raw and Hydrothermally Pretreated Banana Bulbs Biomass in Simultaneous Saccharification and Fermentation Process with Saccharomyces Cerevisiae. Biomass Convers. Biorefinery 2019, 9, 553–563. [Google Scholar] [CrossRef]
- Garriga, M.; Almaraz, M.; Marchiaro, A. Determination of Reducing Sugars in Extracts of Undaria Pinnatifida (Harvey) Algae by UV-Visible Spectrophotometry (DNS Method) Determinación de Azúcares Reduc-tores En Extractos de Alga Undaria Pinnatifida (Harvey) Por Espectrofotometriía UV-Visible (Meétodo DNS). Actas Ing. 2017, 3, 173–179. [Google Scholar]
- Lyngwi, N.A.; Koijam, K.; Sharma, D.; Joshi, S.R. Cultivable bacterial diversity along the altitudinal zonation and vegetation range of tropical Eastern Himalaya. Rev. Biol. Trop. 2013, 61, 467–490. [Google Scholar] [CrossRef]
- Keller, A.G.; Apprill, A.; Lebaron, P.; Robbins, J.; Romano, T.A.; Overton, E.; Rong, Y.; Yuan, R.; Pollara, S.; Whalen, E.K. Characterizing the culturable surface microbiomes of diverse marine animals. FEMS Microbiol. Ecol. 2021, 97, fiab040. [Google Scholar] [CrossRef]
- Jan, B.; Reshi, Z.A.; Mohiddin, F.A. Site and Organ-Specific Culture-Dependent Endophytic Diversity of Crocus sativus L. (Saffron) in Kashmir Himalaya, India. Microb. Ecol. 2021, 83, 989–1006. [Google Scholar] [CrossRef]
- Aires, T.; Serrão, E.A.; Engelen, A.H. Host and Environmental Specificity in Bacterial Communities Associated to Two Highly Invasive Marine Species (Genus Asparagopsis). Front. Microbiol. 2016, 7, 559. [Google Scholar] [CrossRef]
- Tang, J.-C.; Taniguchi, H.; Chu, H.; Zhou, Q.; Nagata, S. Isolation and characterization of alginate-degrading bacteria for disposal of seaweed wastes. Lett. Appl. Microbiol. 2009, 48, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Kizhakkekalam, V.K.; Chakraborty, K. Pharmacological properties of marine macroalgae-associated heterotrophic bacteria. Arch. Microbiol. 2018, 201, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Kai, W.; Peisheng, Y.; Rui, M.; Wenwen, J.; Zongze, S. Diversity of culturable bacteria in deep-sea water from the South Atlantic Ocean. Bioengineered 2017, 8, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Beleneva, I.A.; Zhukova, N.V. Bacterial communities of some brown and red algae from Peter the Great Bay, the Sea of Japan. Microbiology 2006, 75, 348–357. [Google Scholar] [CrossRef]
- Menezes, C.B.; Bonugli-Santos, R.C.; Miqueletto, P.B.; Passarini, M.R.; Silva, C.H.; Justo, M.R.; Leal, R.R.; Fantinatti-Garboggini, F.; Oliveira, V.M.; Berlinck, R.G.; et al. Microbial diversity associated with algae, ascidians and sponges from the north coast of São Paulo state, Brazil. Microbiol. Res. 2010, 165, 466–482. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, R.; Leiva, S. Agar-degrading bacteria isolated from Antarctic macroalgae. Folia Microbiol. 2017, 62, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Ihua, M.W.; Guihéneuf, F.; Mohammed, H.; Margassery, L.M.; Jackson, S.A.; Stengel, D.B.; Clarke, D.J.; Dobson, A.D.W. Microbial Population Changes in Decaying Ascophyllum nodosum Result in Macroalgal-Polysaccharide-Degrading Bacteria with Potential Applicability in Enzyme-Assisted Extraction Technologies. Mar. Drugs 2019, 17, 200. [Google Scholar] [CrossRef]
- Reen, F.J.; Almagro-Moreno, S.; Ussery, D.; Boyd, E.F. The genomic code: Inferring Vibrionaceae niche specialization. Nat. Rev. Genet. 2006, 4, 697–704. [Google Scholar] [CrossRef]
- Dobretsov, S.; Abed, R.M.; Teplitski, M. Mini-review: Inhibition of biofouling by marine microorganisms. Biofouling 2013, 29, 423–441. [Google Scholar] [CrossRef]
- Dobretsov, S.; Dahms, H.-U.; Qian, P.-Y. Inhibition of biofouling by marine microorganisms and their metabolites. Biofouling 2006, 22, 43–54. [Google Scholar] [CrossRef]
- Sinimol, S.; Sarika, A.R.; Nair, A.J. Diversity and antagonistic potential of marine microbes collected from south-west coast of India. 3 Biotech 2015, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Vieira, C.; Engelen, A.; Guentas, L.; Aires, T.; Houlbreque, F.; Gaubert, J.; Serrao, E.; De Clerck, O.; Payri, C. Species Specificity of Bacteria Associated to the Brown Seaweeds Lobophora (Dictyotales, Phaeophyceae) and Their Potential for Induction of Rapid Coral Bleaching in Acropora muricata. Front. Microbiol. 2016, 7, 316. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chen, L.; Zhang, Z.; Wang, X.; Qin, S.; Yan, P. Screening of alginate lyase-excreting microorganisms from the surface of brown algae. AMB Express 2017, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Barbato, M.; Vacchini, V.; Engelen, A.H.; Patania, G.; Mapelli, F.; Borin, S.; Crotti, E. What lies on macroalgal surface: Diversity of polysaccharide degraders in culturable epiphytic bacteria. AMB Express 2022, 12, 98. [Google Scholar] [CrossRef]
- Hollants, J.; Leliaert, F.; De Clerck, O.; Willems, A. What We Can Learn from Sushi: A Review on Sea-weed-Bacterial Associations. FEMS Microbiol. Ecol. 2013, 83, 1–16. [Google Scholar] [CrossRef]
- Bowman, J.P. Bioactive Compound Synthetic Capacity and Ecological Significance of Marine Bacterial Genus. Pseudoalteromonas 2007, 5, 220–241. [Google Scholar] [CrossRef]
- Skerratt, J.H.; Bowman, J.; Hallegraeff, G.; James, S.; Nichols, P.D. Algicidal bacteria associated with blooms of a toxic dinoflagellate in a temperate Australian estuary. Mar. Ecol. Prog. Ser. 2002, 244, 1–15. [Google Scholar] [CrossRef]
- Lee, B.-K.; Katano, T.; Kitamura, S.-I.; Oh, M.-J.; Han, M.-S. Monitoring of Algicidal Bacterium, Alteromonas sp. Strain A14 in Its Application to Natural Cochlodinium Polykrikoides Blooming Seawater Using Fluorescence in Situ Hybridization. J. Microbiol. 2008, 46, 274–282. [Google Scholar] [CrossRef]
- Egan, S.; James, S.; Holmström, C.; Kjelleberg, S. Inhibition of Algal Spore Germination by the Marine Bacterium Pseudoalteromonas Tunicata. FEMS Microbiol. Ecol. 2001, 35, 67–73. [Google Scholar] [CrossRef]
- Dobretsov, S.V.; Qian, P.-Y. Effect of Bacteria Associated with the Green Alga Ulva reticulata on Marine Micro- and Macrofouling. Biofouling 2002, 18, 217–228. [Google Scholar] [CrossRef]
- Patel, P.; Callow, M.E.; Joint, I.; Callow, J.A. Specificity in the settlement-modifying response of bacterial biofilms towards zoospores of the marine alga Enteromorpha. Environ. Microbiol. 2003, 5, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Kanagasabhapathy, M.; Sasaki, H.; Haldar, S.; Yamasaki, S.; Nagata, S. Antibacterial activities of marine epibiotic bacteria isolated from brown algae of Japan. Ann. Microbiol. 2006, 56, 167–173. [Google Scholar] [CrossRef]
- Egan, S.; Thomas, T.; Kjelleberg, S. Unlocking the diversity and biotechnological potential of marine surface associated microbial communities. Curr. Opin. Microbiol. 2008, 11, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Matz, C.; Webb, J.; Schupp, P.J.; Phang, S.Y.; Penesyan, A.; Egan, S.; Steinberg, P.; Kjelleberg, S. Marine Biofilm Bacteria Evade Eukaryotic Predation by Targeted Chemical Defense. PLoS ONE 2008, 3, e2744. [Google Scholar] [CrossRef] [PubMed]
- Ballestriero, F.; Thomas, T.; Burke, C.; Egan, S.; Kjelleberg, S. Identification of Compounds with Bioactivity against the Nematode Caenorhabditis elegans by a Screen Based on the Functional Genomics of the Marine Bacterium Pseudoalteromonas tunicata D2. Appl. Environ. Microbiol. 2010, 76, 5710–5717. [Google Scholar] [CrossRef]
- Prakash, O.; Nimonkar, Y.; Shouche, Y.S. Practice and prospects of microbial preservation. FEMS Microbiol. Lett. 2012, 339, 1–9. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M.; Spröer, C.; Klenk, H.-P. When should a DDH experiment be mandatory in microbial taxonomy? Arch. Microbiol. 2013, 195, 413–418. [Google Scholar] [CrossRef]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; Da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.-W.; De Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef]
- Verma, A.; Ojha, A.K.; Dastager, S.G.; Natarajan, R.; Mayilraj, S.; Krishnamurthi, S. Domibacillus mangrovi sp. nov. and Domibacillus epiphyticus sp. nov., isolated from marine habitats of the central west coast of India. Int. J. Syst. Evol. Microbiol. 2017, 67, 3063–3070. [Google Scholar] [CrossRef]
- Verma, A.; Ojha, A.K.; Kumari, P.; Sundharam, S.S.; Mayilraj, S.; Krishnamurthi, S.; Mual, P. Luteimonas padinae sp. nov., an epiphytic bacterium isolated from an intertidal macroalga. Int. J. Syst. Evol. Microbiol. 2016, 66, 5444–5451. [Google Scholar] [CrossRef]
- Brunet, M.; Le Duff, N.; Barbeyron, T.; Thomas, F. Consuming fresh macroalgae induces specific catabolic pathways, stress reactions and Type IX secretion in marine flavobacterial pioneer degraders. ISME J. 2022, 16, 2027–2039. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, C.; Hidalgo, C.; Zapata, M.; Jeison, D.; Riquelme, C.; Rivas, M. Use of Cellulolytic Marine Bacteria for Enzymatic Pretreatment in Microalgal Biogas Production. Appl. Environ. Microbiol. 2014, 80, 4199–4206. [Google Scholar] [CrossRef] [PubMed]
- Naik, M.M.; Dubey, S.K. Marine Pollution and Microbial Remediation; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar] [CrossRef]
- Martin, M.; Barbeyron, T.; Martin, R.; Portetelle, D.; Michel, G.; Vandenbol, M. The Cultivable Surface Microbiota of the Brown Alga Ascophyllum nodosum is Enriched in Macroalgal-Polysaccharide-Degrading Bacteria. Front. Microbiol. 2015, 6, 1487. [Google Scholar] [CrossRef] [PubMed]
- Imran; Poduval, P.B.; Ghadi, S.C. Bacterial Degradation of Algal Polysaccharides in Marine Ecosystem. In Marine Pollution and Microbial Remediation; Springer: Singapore, 2016; pp. 189–203. ISBN 9789811010446. [Google Scholar]
- Comba-González, N.B.; Ruiz-Toquica, J.S.; Lopez-Kleine, L.; Montoya-Castano, D. Epiphytic Bacteria of Macroalgae of the Genus Ulva and their Potential in Producing Enzymes Having Biotechnological Interest. J. Mar. Biol. Oceanogr. 2016, 5, 2–9. [Google Scholar] [CrossRef]
- Gebbie, L.; Dam, T.T.; Ainscough, R.; Palfreyman, R.; Cao, L.; Harrison, M.; O’Hara, I.; Speight, R. A snapshot of microbial diversity and function in an undisturbed sugarcane bagasse pile. BMC Biotechnol. 2020, 20, 12. [Google Scholar] [CrossRef]
- Laopaiboon, P.; Thani, A.; Leelavatcharamas, V.; Laopaiboon, L. Acid hydrolysis of sugarcane bagasse for lactic acid production. Bioresour. Technol. 2010, 101, 1036–1043. [Google Scholar] [CrossRef]
- Mesa, L.; González, E.; Cara, C.; Castro, E.; Mussatto, S. The effect of organosolv pretreatment variables on enzymatic hydrolysis of sugarcane bagasse. Chem. Eng. J. 2011, 168, 1157–1162. [Google Scholar] [CrossRef]
- Guilherme, A.A.; Dantas, P.V.F.; Santos, E.; Fernandes, F.; Macedo, G.R. Evaluation of composition, characterization and enzymatic hydrolysis of pretreated sugar cane bagasse. Braz. J. Chem. Eng. 2015, 32, 23–33. [Google Scholar] [CrossRef]
- Gaur, R.; Tiwari, S.; Pathak, P. Sugarcane Baggase Agro-waste Material Used for Renewable Cellulase Production from Streptococcus and Bacillus sp. Res. J. Microbiol. 2017, 12, 255–265. [Google Scholar] [CrossRef][Green Version]
- Nargotra, P.; Vaid, S.; Bajaj, B.K. Cellulase Production from Bacillus subtilis SV1 and Its Application Potential for Saccharification of Ionic Liquid Pretreated Pine Needle Biomass under One Pot Consolidated Bioprocess. Fermentation 2016, 2, 19. [Google Scholar] [CrossRef]
- Talia, P.; Sede, S.M.; Campos, E.; Rorig, M.; Principi, D.; Tosto, D.; Hopp, H.E.; Grasso, D.; Cataldi, A. Biodiversity characterization of cellulolytic bacteria present on native Chaco soil by comparison of ribosomal RNA genes. Res. Microbiol. 2012, 163, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Bharathiraja, S.; Suriya, J.; Krishnan, M.; Manivasagan, P.; Kim, S.-K. Production of Enzymes from Agricultural Wastes and Their Potential Industrial Applications. In Advances in Food and Nutrition Research; Elsevier BV: Amsterdam, The Netherlands, 2017; Volume 80, pp. 125–148. [Google Scholar]
- Taheri, M.E.; Salimi, E.; Saragas, K.; Novakovic, J.; Barampouti, E.M.; Mai, S.; Malamis, D.; Moustakas, K.; Loizidou, M. Effect of pretreatment techniques on enzymatic hydrolysis of food waste. Biomass-Convers. Biorefinery 2020, 11, 219–226. [Google Scholar] [CrossRef]
- Kunamneni, A.; Singh, S. Response surface optimization of enzymatic hydrolysis of maize starch for higher glucose production. Biochem. Eng. J. 2005, 27, 179–190. [Google Scholar] [CrossRef]
- Bohra, V.; Dafale, N.A.; Hathi, Z.; Purohit, H.J. Genomic annotation and validation of bacterial consortium NDMC-1 for enhanced degradation of sugarcane bagasse. Ann. Microbiol. 2019, 69, 695–711. [Google Scholar] [CrossRef]
- Di Marco, E.; Soraire, P.M.; Romero, C.M.; Villegas, L.B.; Martínez, M.A. Raw sugarcane bagasse as carbon source for xylanase production by Paenibacillus species: A potential degrader of agricultural wastes. Environ. Sci. Pollut. Res. 2017, 24, 19057–19067. [Google Scholar] [CrossRef] [PubMed]
- Delabona, P.D.S.; Pirota, R.D.B.; Codima, C.A.; Tremacoldi, C.R.; Rodrigues, A.; Farinas, C.S. Using Amazon forest fungi and agricultural residues as a strategy to produce cellulolytic enzymes. Biomass-Bioenergy 2012, 37, 243–250. [Google Scholar] [CrossRef]
- Dos Santos, B.S.L.; Gomes, A.F.S.; Franciscon, E.G.; Oliveira, J.; Baffi, M.A. Thermotolerant and mesophylic fungi from sugarcane bagasse and their prospection for biomass-degrading enzyme production. Braz. J. Microbiol. 2015, 46, 903–910. [Google Scholar] [CrossRef]
- Popper, Z.A.; Michel, G.; Hervé, C.; Domozych, D.S.; Willats, W.G.; Tuohy, M.G.; Kloareg, B.; Stengel, D.B. Evolution and Diversity of Plant Cell Walls: From Algae to Flowering Plants. Annu. Rev. Plant Biol. 2011, 62, 567–590. [Google Scholar] [CrossRef]

 ) and raw sugarcane bagasse (
) and raw sugarcane bagasse ( ) are appropriately highlighted. Novel strains at genus and species level (published and putative) are highlighted with bold and yellow background. Gaps and ambiguous bases were removed from the final data sets. The final tree was visualized and edited in iTOL server.
) and raw sugarcane bagasse () are appropriately highlighted. Novel strains at genus and species level (published and putative) are highlighted with bold and yellow background. Gaps and ambiguous bases were removed from the final data sets. The final tree was visualized and edited in iTOL server.
) are appropriately highlighted. Novel strains at genus and species level (published and putative) are highlighted with bold and yellow background. Gaps and ambiguous bases were removed from the final data sets. The final tree was visualized and edited in iTOL server.
) and raw sugarcane bagasse () are appropriately highlighted. Novel strains at genus and species level (published and putative) are highlighted with bold and yellow background. Gaps and ambiguous bases were removed from the final data sets. The final tree was visualized and edited in iTOL server.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. | Sampling Location/GPS Location | Macroalgae/Seaweed |
|---|---|---|
| 1 | Malwan (Maharashtra) 16°03.711′ N, 073°27.329′ E | GAMAL (Ulva sp.), MBA (Sargassum polycystum), MRA (Pterocladia musciformis) |
| 2 | Anjuna (Goa) 15°34.729′ N, 073°44.303′ E | LLAB (Sargassum polycystum), SAB (Dictyota sp.) |
| 3 | Cado-De-Rama (Goa), 15°06.313′ N, 073°55.437′ E | CDRSL (Padina antillarum) |
| 4 | Kunkeshwar (Maharashtra) 16°20.014′ N, 073°23.505′ E | LLKUN (Sargassum polycystum) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, P.; Verma, A.; Sundharam, S.S.; Ojha, A.K.; Krishnamurthi, S. Exploring Diversity and Polymer Degrading Potential of Epiphytic Bacteria Isolated from Marine Macroalgae. Microorganisms 2022, 10, 2513. https://doi.org/10.3390/microorganisms10122513
Kumar P, Verma A, Sundharam SS, Ojha AK, Krishnamurthi S. Exploring Diversity and Polymer Degrading Potential of Epiphytic Bacteria Isolated from Marine Macroalgae. Microorganisms. 2022; 10(12):2513. https://doi.org/10.3390/microorganisms10122513
Chicago/Turabian StyleKumar, Pravin, Ashish Verma, Shiva S. Sundharam, Anup Kumar Ojha, and Srinivasan Krishnamurthi. 2022. "Exploring Diversity and Polymer Degrading Potential of Epiphytic Bacteria Isolated from Marine Macroalgae" Microorganisms 10, no. 12: 2513. https://doi.org/10.3390/microorganisms10122513
APA StyleKumar, P., Verma, A., Sundharam, S. S., Ojha, A. K., & Krishnamurthi, S. (2022). Exploring Diversity and Polymer Degrading Potential of Epiphytic Bacteria Isolated from Marine Macroalgae. Microorganisms, 10(12), 2513. https://doi.org/10.3390/microorganisms10122513