
Assessment of Hydrocarbon Degradation Potential in Microbial Communities in Arctic Sea Ice




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Mesocosm Setup
2.2. DNA Extraction
2.3. Metagenome Sequencing
2.4. Bioinformatical Analysis
2.4.1. Taxonomic Profiling of the Prokaryotic Community
2.4.2. Construction of Metagenome-Assembled Genomes
2.4.3. Detection of Hydrocarbon Degradation Genes
3. Results
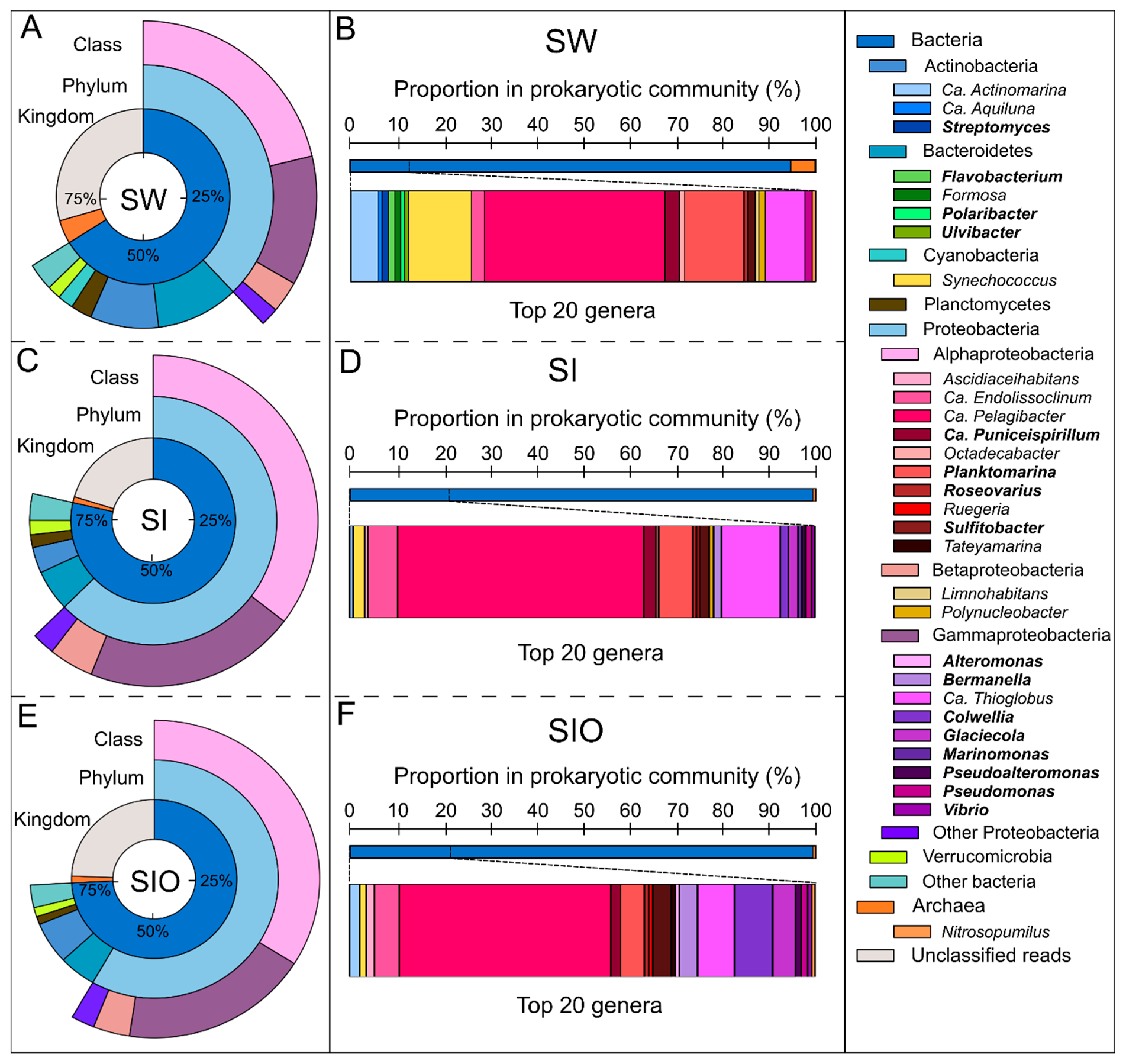
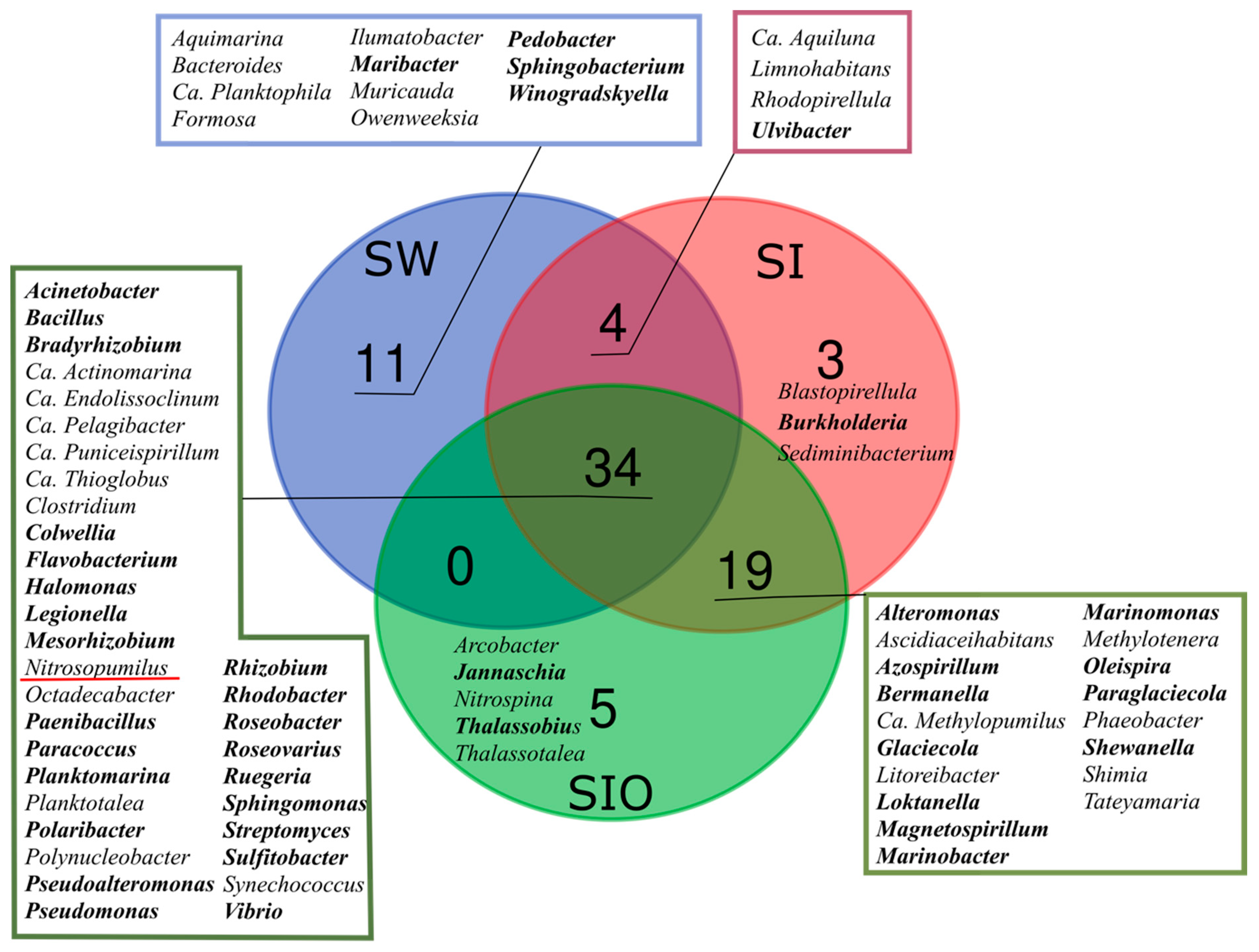
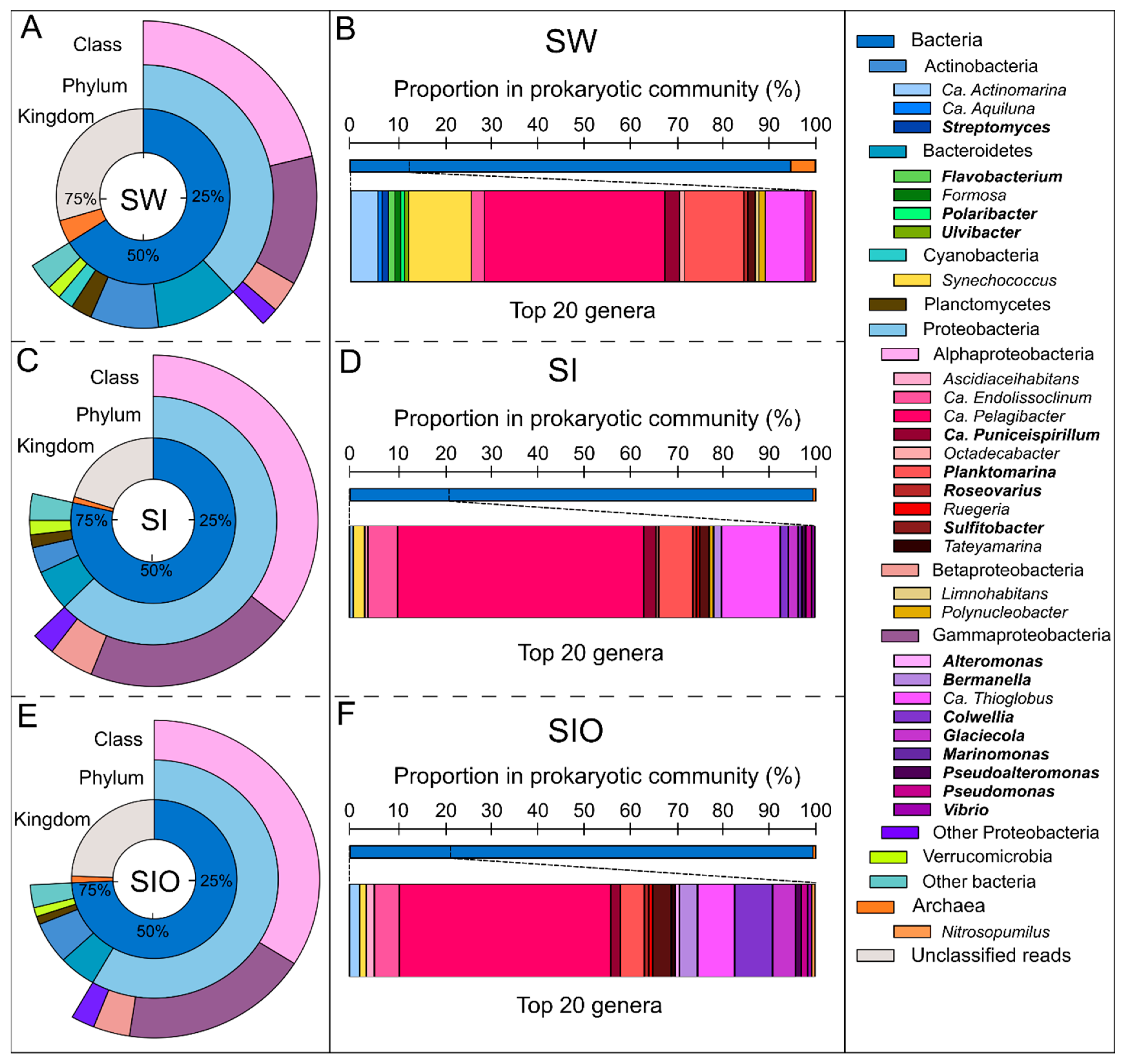
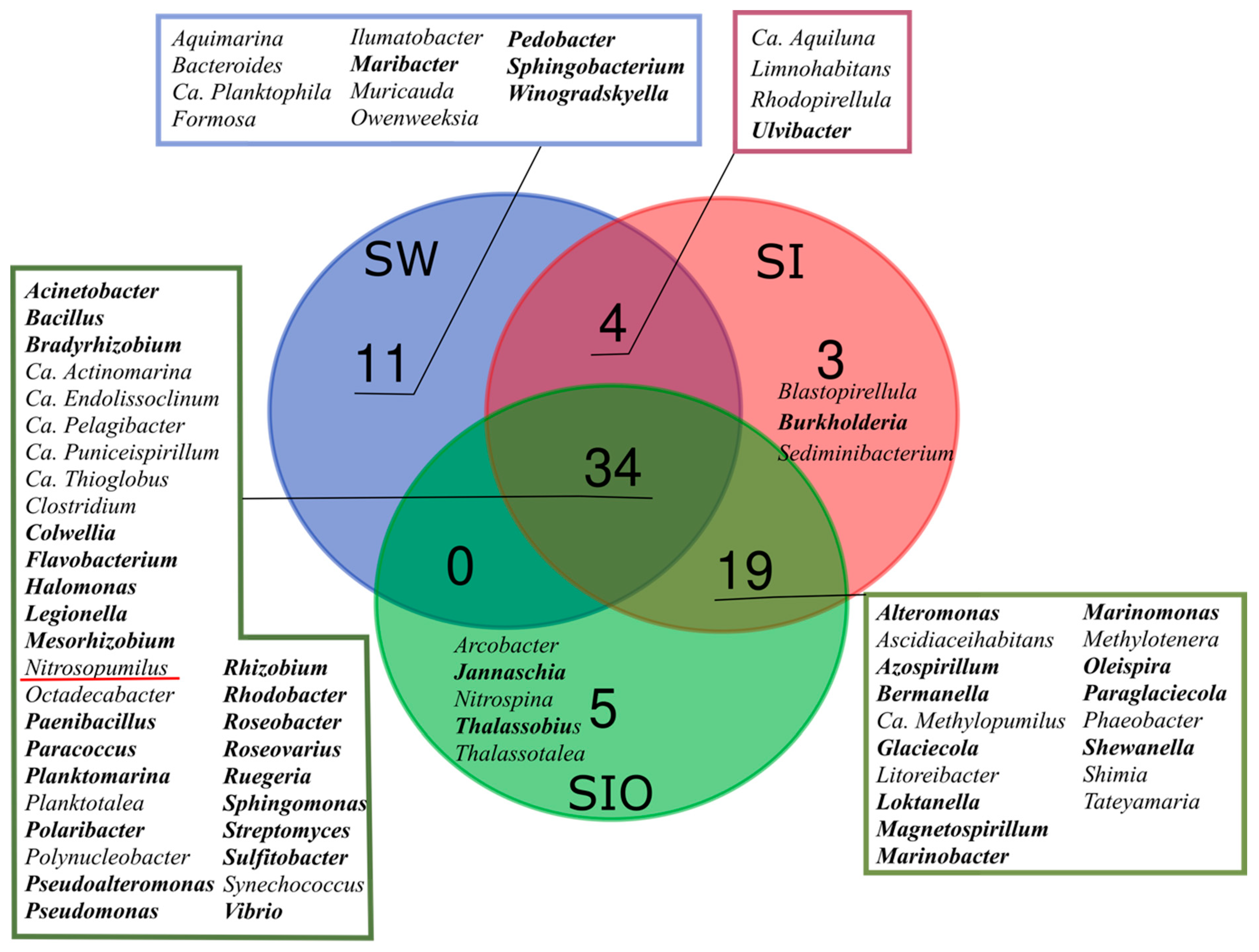
3.1. The Microbial Community Structure in Seawater and Sea Ice
3.2. Proportion of Genera Containing Oil Hydrocarbon-Degrading Organisms in Metagenomes
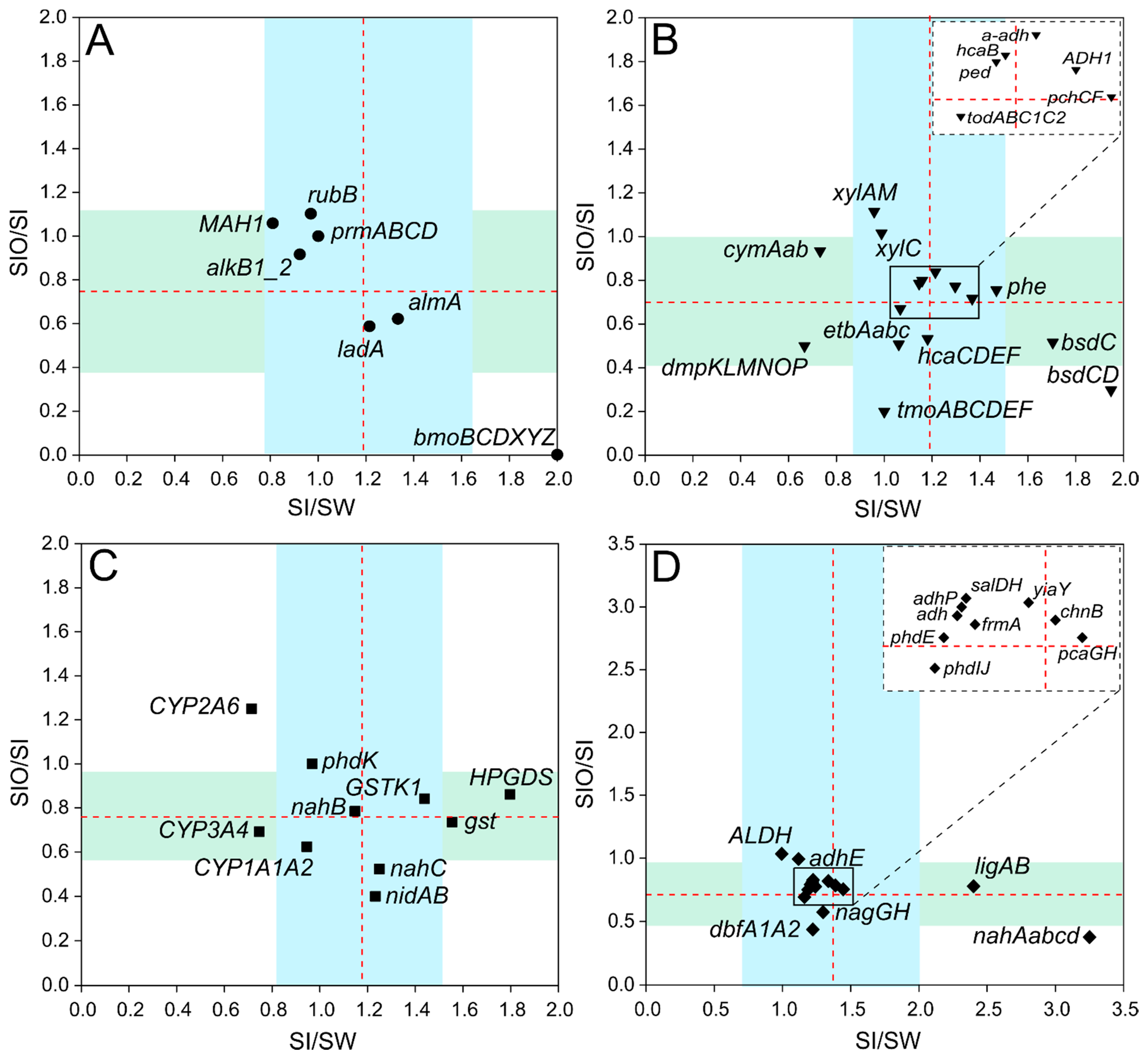
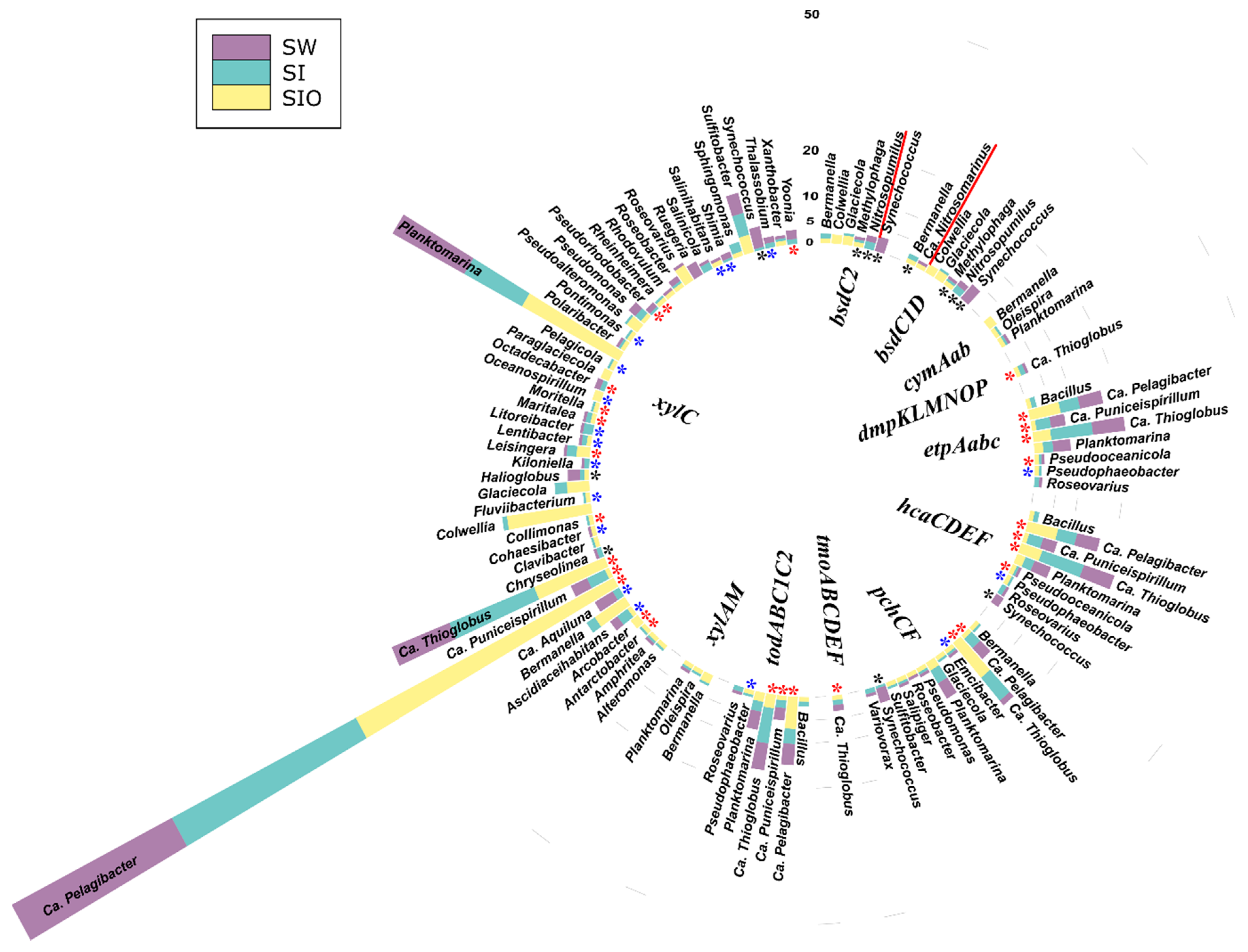
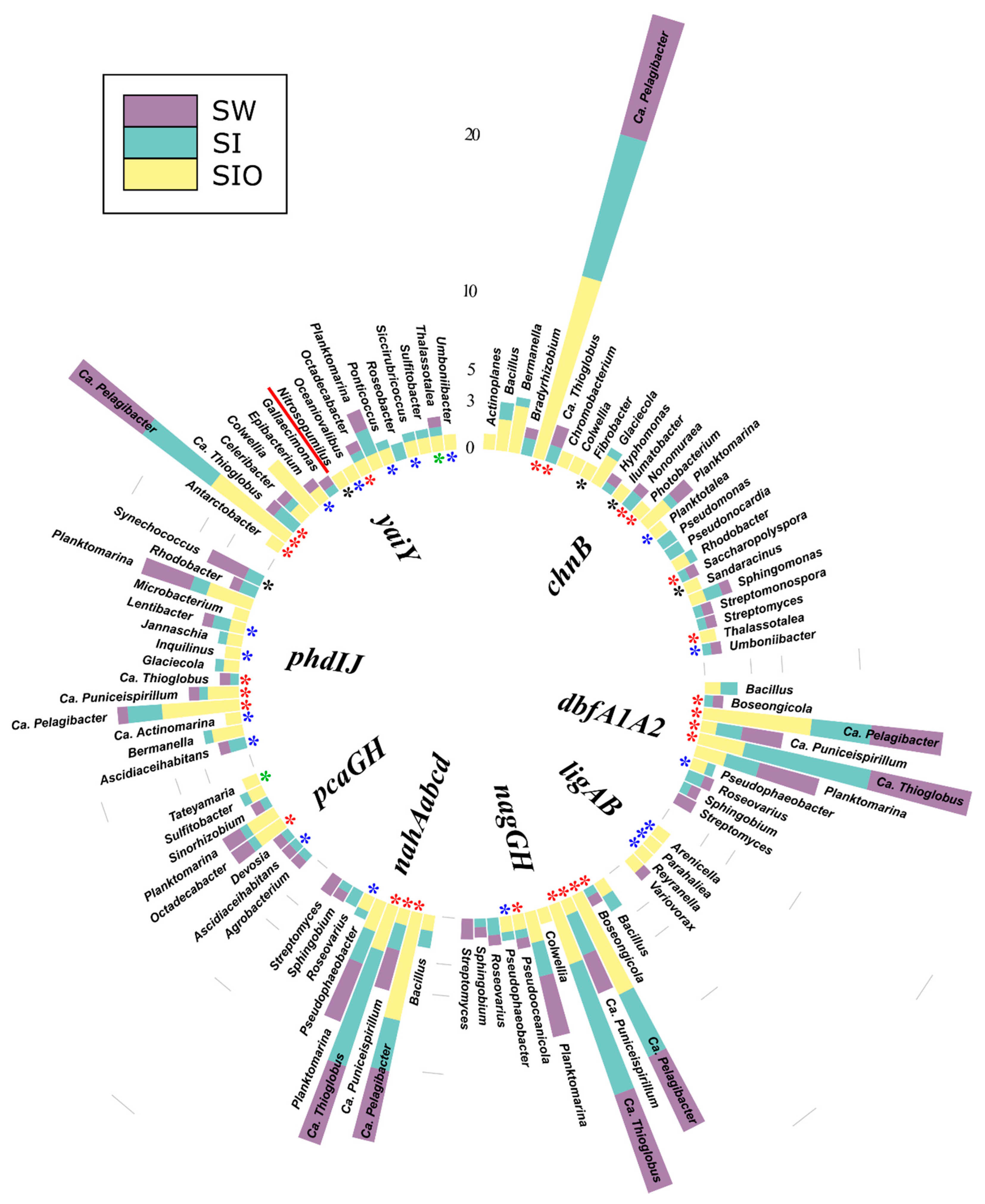
3.3. The Abundance of Genes Associated with Hydrocarbon Degradation in Metagenomes
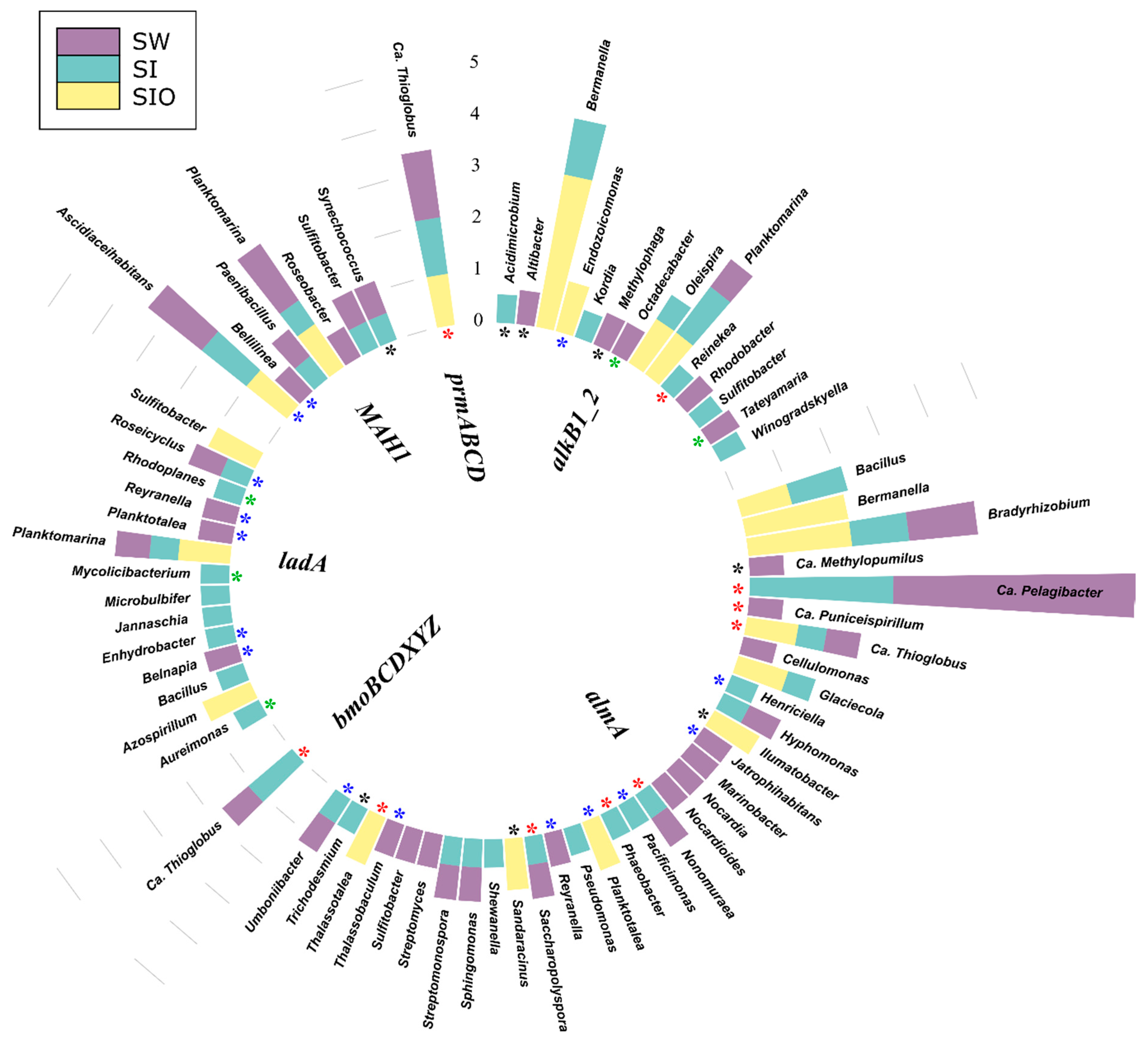
3.4. Association of Hydrocarbon Degradation Genes with Community Phylogenic Composition
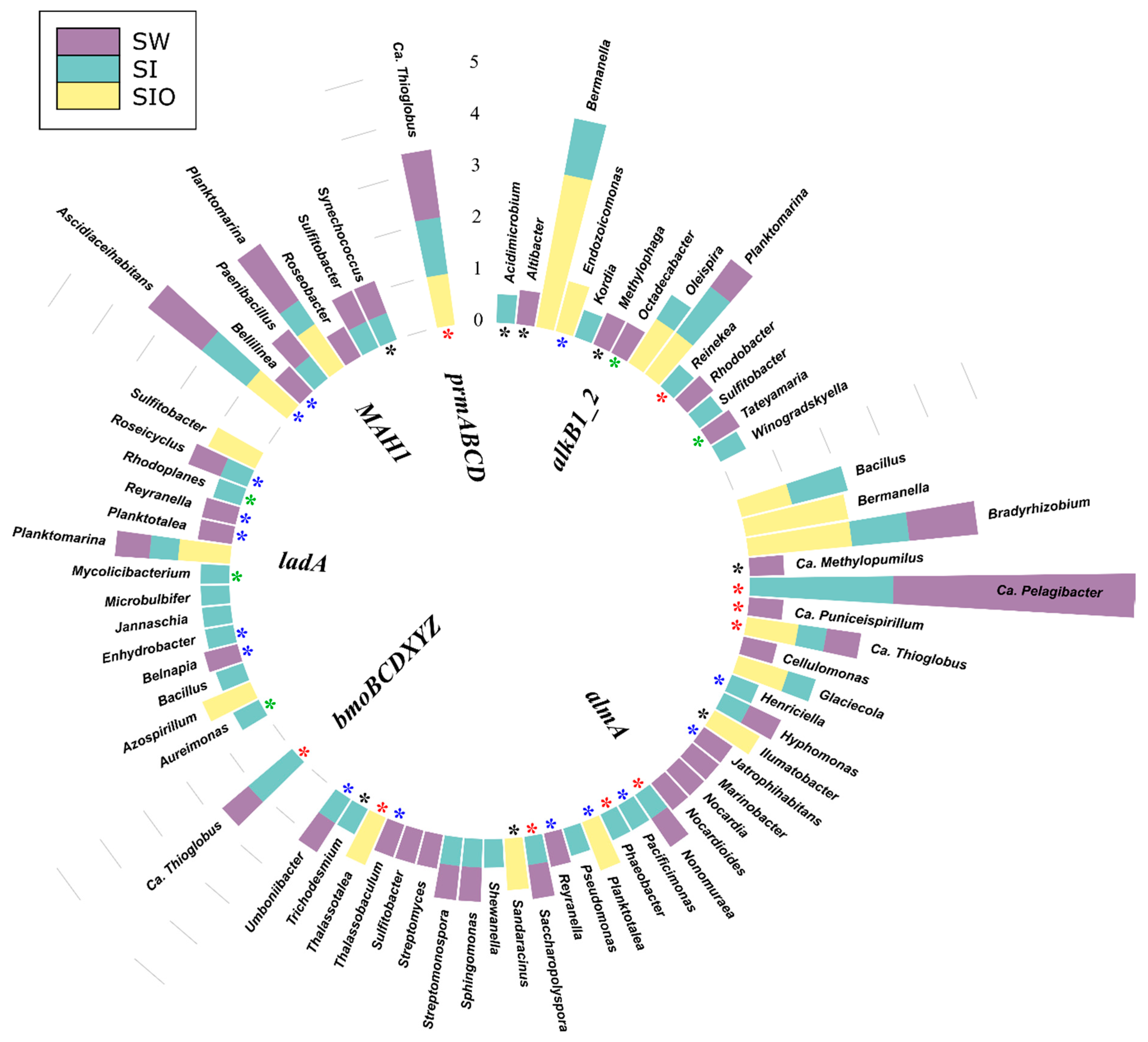
3.4.1. Associations of Alkane Degradation Genes
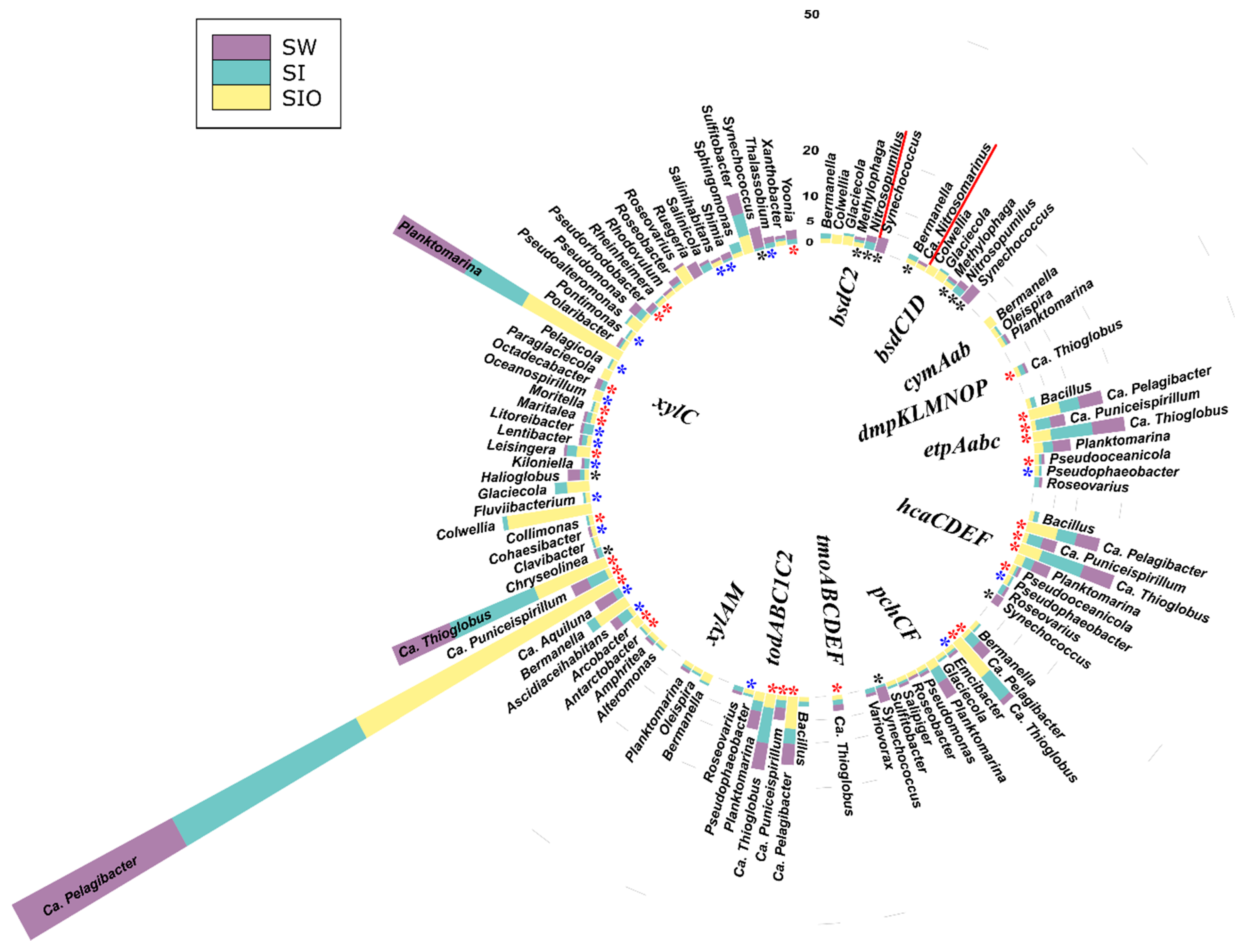
3.4.2. Associations of the Monoaromatic Hydrocarbon Degradation Genes
3.4.3. Associations of the Polyaromatic Hydrocarbon Degradation Genes
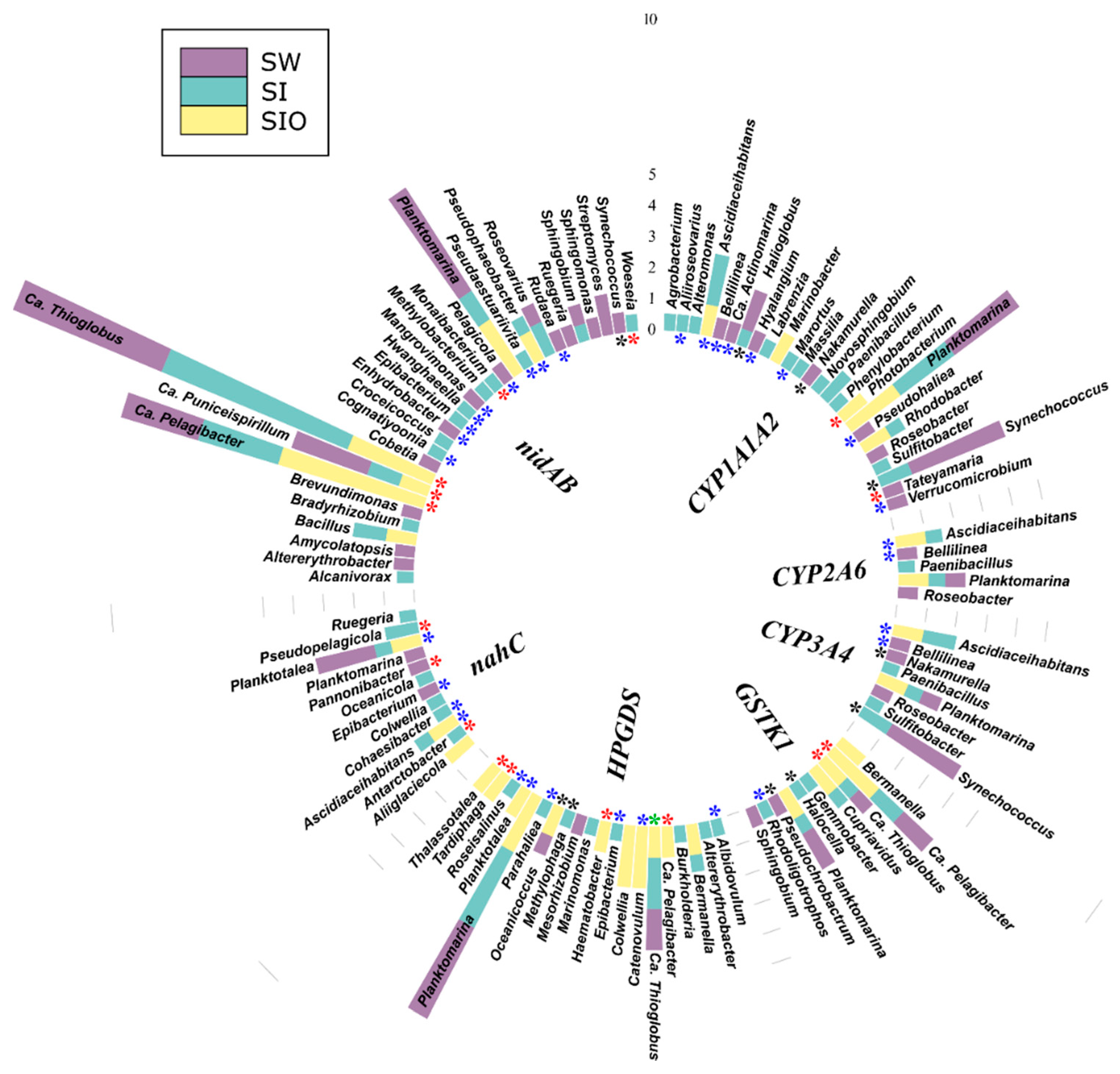
3.4.4. Associations of the Genes Participating in the Degradation of Various Hydrocarbons
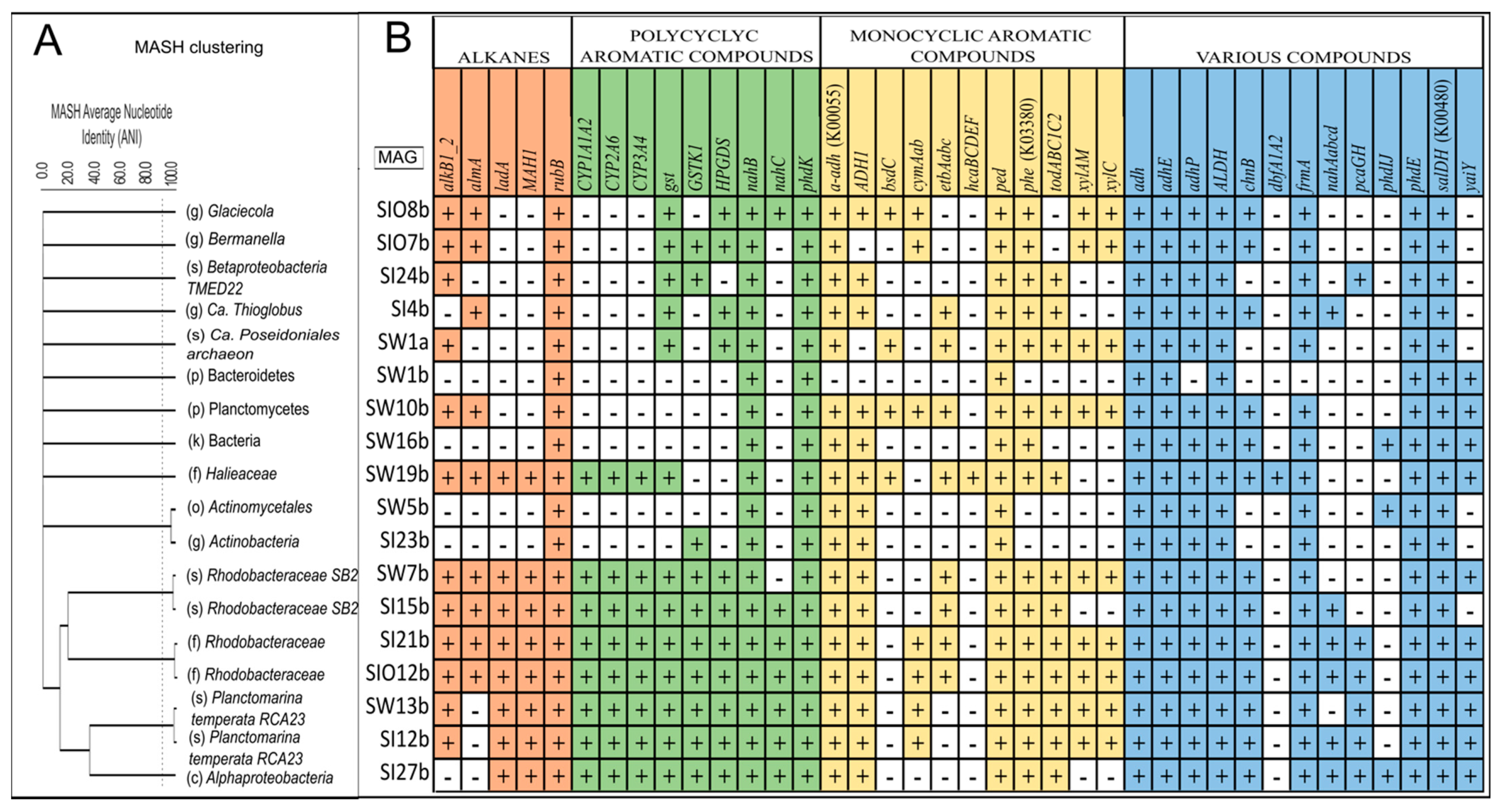
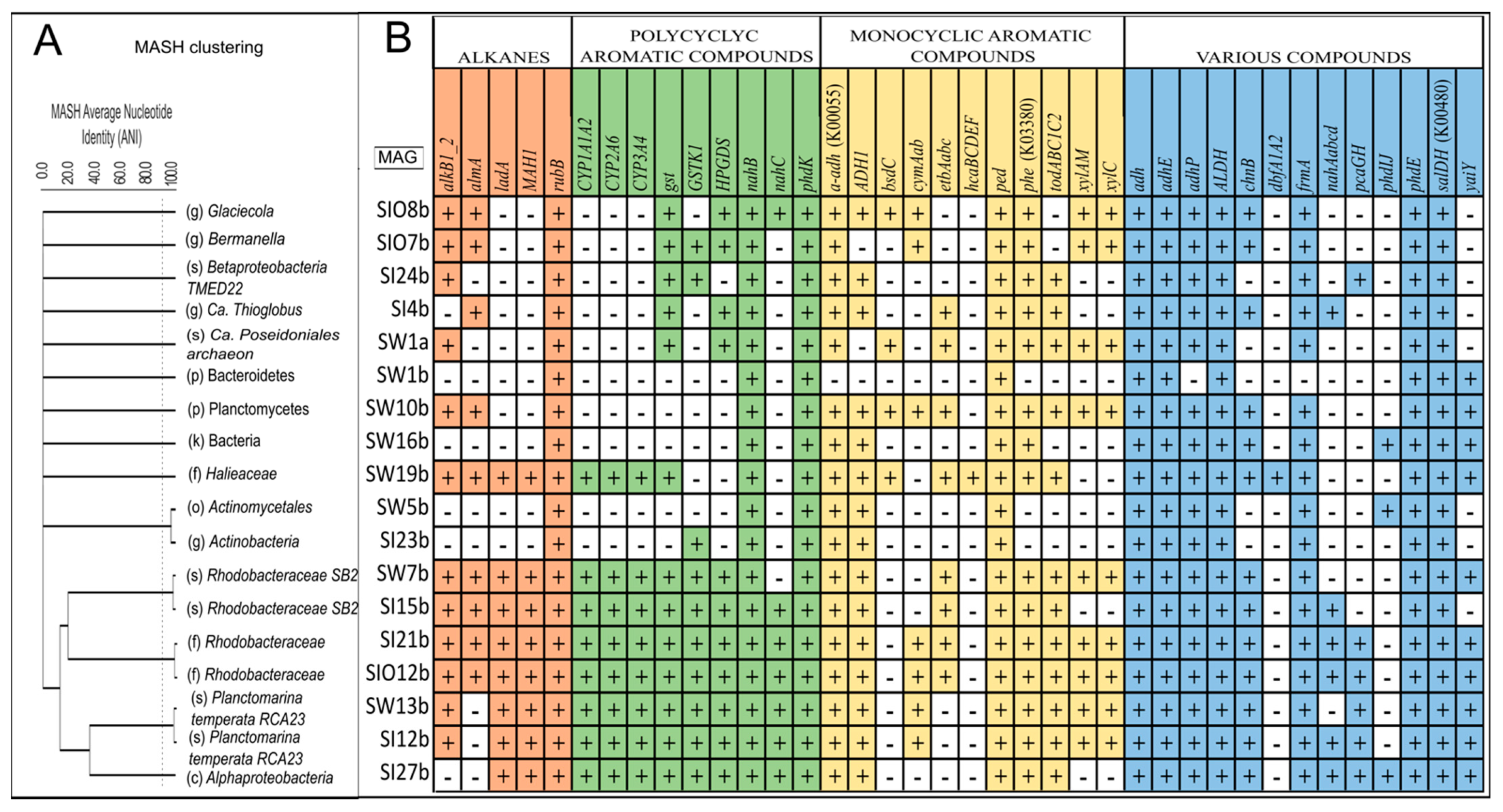
3.5. Metagenome-Assembled Genomes
3.5.1. Taxonomic Affiliation of MAGs
3.5.2. The Genes Related to Hydrocarbon Degradation in Metagenome-Assembled Genomes
4. Discussion
4.1. Prokaryotic Community in Arctic Seawater and Sea Ice
4.2. Hydrocarbon-Degrading Organisms in the Seawater and Sea Ice Communities
4.3. Genetic Potential of Prokaryotic Community to Degrade Crude Oil Compounds in Sea Ice
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sunagawa, S.; Acinas, S.G.; Bork, P.; Bowler, C.; Acinas, S.G.; Babin, M.; Bork, P.; Boss, E.; Bowler, C.; Cochrane, G.; et al. Tara Oceans: Towards global ocean ecosystems biology. Nat. Rev. Genet. 2020, 18, 428–445. [Google Scholar] [CrossRef]
- Ławniczak, Ł.; Woźniak-Karczewska, M.; Loibner, A.P.; Heipieper, H.J.; Chrzanowski, Ł. Microbial Degradation of Hydrocarbons—Basic Principles for Bioremediation: A Review. Molecules 2020, 25, 856. [Google Scholar] [CrossRef] [Green Version]
- Hazen, T.C.; Prince, R.; Mahmoudi, N. Marine Oil Biodegradation. Environ. Sci. Technol. 2016, 50, 2121–2129. [Google Scholar] [CrossRef]
- Hassanshahian, M.; Amirinejad, N.; Behzadi, M.A. Crude oil pollution and biodegradation at the Persian Gulf: A comprehensive and review study. J. Environ. Health Sci. Eng. 2020, 18, 1415–1435. [Google Scholar] [CrossRef]
- Gerdes, B.; Brinkmeyer, R.; Dieckmann, G.; Helmke, E. Influence of crude oil on changes of bacterial communities in Arctic sea-ice. FEMS Microbiol. Ecol. 2005, 53, 129–139. [Google Scholar] [CrossRef]
- Boylan, B.M. Increased maritime traffic in the Arctic: Implications for governance of Arctic sea routes. Mar. Policy 2021, 131, 104566. [Google Scholar] [CrossRef]
- Beyer, J.; Trannum, H.C.; Bakke, T.; Hodson, P.V.; Collier, T.K. Environmental effects of the Deepwater Horizon oil spill: A review. Mar. Pollut. Bull. 2016, 110, 28–51. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, A. Oil pollution in the North Sea: The impact of governance measures on oil pollution over several decades. Hydrobiologia 2018, 845, 109–127. [Google Scholar] [CrossRef] [Green Version]
- Ranellone, R.T.; Tukaew, P.; Shi, X.; Rangwala, A.S. Ignitability of crude oil and its oil-in-water products at arctic temperature. Mar. Pollut. Bull. 2017, 115, 261–265. [Google Scholar] [CrossRef]
- Wilkinson, J.; Beegle-Krause, C.; Evers, K.-U.; Hughes, N.; Lewis, A.; Reed, M.; Wadhams, P. Oil spill response capabilities and technologies for ice-covered Arctic marine waters: A review of recent developments and established practices. Ambio 2017, 46, 423–441. [Google Scholar] [CrossRef] [Green Version]
- Van Stempvoort, D.; Biggar, K. Potential for bioremediation of petroleum hydrocarbons in groundwater under cold climate conditions: A review. Cold Reg. Sci. Technol. 2008, 53, 16–41. [Google Scholar] [CrossRef]
- Guerin, T.F. A safe, efficient and cost effective process for removing petroleum hydrocarbons from a highly heterogeneous and relatively inaccessible shoreline. J. Environ. Manag. 2015, 162, 190–198. [Google Scholar] [CrossRef]
- Thomas, G.; Brant, J.; Campo, P.; Clark, D.; Coulon, F.; Gregson, B.; McGenity, T.; McKew, B. Effects of Dispersants and Biosurfactants on Crude-Oil Biodegradation and Bacterial Community Succession. Microorganisms 2021, 9, 1200. [Google Scholar] [CrossRef]
- Amann, R.I.; Ludwig, W.; Schleifer, K.H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 1995, 59, 143–169. [Google Scholar] [CrossRef]
- Riesenfeld, C.S.; Schloss, P.D.; Handelsman, J. Metagenomics: Genomic Analysis of Microbial Communities. Annu. Rev. Genet. 2004, 38, 525–552. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Zhang, W.; Ding, W.; Wang, M.; Fan, S.; Yang, B.; Mcminn, A.; Wang, M.; Xie, B.-B.; Qin, Q.-L.; et al. Structure and function of the Arctic and Antarctic marine microbiota as revealed by metagenomics. Microbiome 2020, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [Green Version]
- McFarlin, K.; Perkins, M.J.; Field, J.A.; Leigh, M.B. Biodegradation of Crude Oil and Corexit 9500 in Arctic Seawater. Front. Microbiol. 2018, 9, 1788. [Google Scholar] [CrossRef] [Green Version]
- Vergeynst, L.; Greer, C.W.; Mosbech, A.; Gustavson, K.; Meire, L.; Poulsen, K.G.; Christensen, J.H. Biodegradation, Photo-oxidation, and Dissolution of Petroleum Compounds in an Arctic Fjord during Summer. Environ. Sci. Technol. 2019, 53, 12197–12206. [Google Scholar] [CrossRef]
- Ribicic, D.; Netzer, R.; Hazen, T.; Techtmann, S.M.; Drabløs, F.; Brakstad, O.G. Microbial community and metagenome dynamics during biodegradation of dispersed oil reveals potential key-players in cold Norwegian seawater. Mar. Pollut. Bull. 2018, 129, 370–378. [Google Scholar] [CrossRef] [Green Version]
- De Sousa, S.T.P.; Cabral, L.; Lacerda-Júnior, G.V.; Noronha, M.F.; Ottoni, J.R.; Sartoratto, A.; De Oliveira, V.M. Exploring the genetic potential of a fosmid metagenomic library from an oil-impacted mangrove sediment for metabolism of aromatic compounds. Ecotoxicol. Environ. Saf. 2020, 189, 109974. [Google Scholar] [CrossRef]
- Hidalgo, K.J.; Sierra-Garcia, I.N.; Dellagnezze, B.M.; De Oliveira, V.M. Metagenomic Insights Into the Mechanisms for Biodegradation of Polycyclic Aromatic Hydrocarbons in the Oil Supply Chain. Front. Microbiol. 2020, 11, 561506. [Google Scholar] [CrossRef] [PubMed]
- Fingas, M.; Hollebone, B. Review of behaviour of oil in freezing environments. Mar. Pollut. Bull. 2003, 47, 333–340. [Google Scholar] [CrossRef]
- Oggier, M.; Eicken, H.; Wilkinson, J.; Petrich, C.; O’Sadnick, M. Crude oil migration in sea-ice: Laboratory studies of constraints on oil mobilization and seasonal evolution. Cold Reg. Sci. Technol. 2019, 174, 102924. [Google Scholar] [CrossRef]
- Yergeau, E.; Michel, C.; Tremblay, J.; Niemi, A.; King, T.L.; Wyglinski, J.; Lee, K.; Greer, C.W. Metagenomic survey of the taxonomic and functional microbial communities of seawater and sea ice from the Canadian Arctic. Sci. Rep. 2017, 7, srep42242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boccadoro, C.; Krolicka, A.; Receveur, J.; Aeppli, C.; Le Floch, S. Microbial community response and migration of petroleum compounds during a sea-ice oil spill experiment in Svalbard. Mar. Environ. Res. 2018, 142, 214–233. [Google Scholar] [CrossRef] [PubMed]
- Brakstad, O.G.; Nonstad, I.; Faksness, L.-G.; Brandvik, P.J. Responses of Microbial Communities in Arctic Sea Ice After Contamination by Crude Petroleum Oil. Microb. Ecol. 2007, 55, 540–552. [Google Scholar] [CrossRef]
- Lofthus, S.; Bakke, I.; Greer, C.W.; Brakstad, O.G. Biodegradation of weathered crude oil by microbial communities in solid and melted sea ice. Mar. Pollut. Bull. 2021, 172, 112823. [Google Scholar] [CrossRef]
- Krolicka, A.; Boccadoro, C.; Nilsen, M.M.; Demir-Hilton, E.; Birch, J.; Preston, C.; Scholin, C.; Baussant, T. Identification of microbial key-indicators of oil contamination at sea through tracking of oil biotransformation: An Arctic field and laboratory study. Sci. Total Environ. 2019, 696, 133715. [Google Scholar] [CrossRef]
- Yakimov, A.M.; Timmis, K.N.; Golyshin, P.N. Obligate oil-degrading marine bacteria. Curr. Opin. Biotechnol. 2007, 18, 257–266. [Google Scholar] [CrossRef]
- Kube, M.; Chernikova, T.N.; Al-Ramahi, Y.; Golyshin, P.N. Genome sequence and functional genomic analysis of the oil-degrading bacterium Oleispira antarctica. Nat. Commun. 2013, 4, 2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S. Babraham Bioinformatics-FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 2 October 2021).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Rodriguez, R.L.M.; Konstantinidis, K.T. Nonpareil: A redundancy-based approach to assess the level of coverage in metagenomic datasets. Bioinformatics 2013, 30, 629–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzel, P.; Ng, K.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nõlvak, H.; Dang, N.P.; Truu, M.; Peeb, A.; Tiirik, K.; O’Sadnick, M.; Truu, J. Microbial Community Dynamics during Biodegradation of Crude Oil and Its Response to Biostimulation in Svalbard Seawater at Low Temperature. Microorganisms 2021, 9, 2425. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. dRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39 (Suppl. 2), W29–W37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Shao, Z. Diversity of flavin-binding monooxygenase genes (almA) in marine bacteria capable of degradation long-chain alkanes. FEMS Microbiol. Ecol. 2012, 80, 523–533. [Google Scholar] [CrossRef] [Green Version]
- Nayfach, S.; Pollard, K.S. Average genome size estimation improves comparative metagenomics and sheds light on the functional ecology of the human microbiome. Genome Biol. 2015, 16, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Boetius, A.; Anesio, A.M.; Deming, J.W.; Mikucki, J.A.; Rapp, J. Microbial ecology of the cryosphere: Sea ice and glacial habitats. Nat. Rev. Genet. 2015, 13, 677–690. [Google Scholar] [CrossRef]
- Amiraux, R.; Rontani, J.-F.; Armougom, F.; Frouin, E.; Babin, M.; Artigue, L.; Bonin, P. Bacterial diversity and lipid biomarkers in sea ice and sinking particulate organic material during the melt season in the Canadian Arctic. Elem. Sci. Anthr. 2021, 9. [Google Scholar] [CrossRef]
- Zhao, X.; Schwartz, C.L.; Pierson, J.; Giovannoni, S.J.; McIntosh, J.R.; Nicastro, D. Three-Dimensional Structure of the Ultraoligotrophic Marine Bacterium “Candidatus Pelagibacter ubique”. Appl. Environ. Microbiol. 2017, 83, e02807-16. [Google Scholar] [CrossRef] [Green Version]
- Shi, K.; Zhang, Q.; Xue, J.; Chen, X.; Chen, Y.; Qiao, Y.; Yang, Y.; Sun, J. Study on the degradation performance and bacterial community of bioaugmentation in petroleum-pollution seawater. J. Environ. Chem. Eng. 2020, 8, 103900. [Google Scholar] [CrossRef]
- Zwirglmaier, K.; Jardillier, L.; Ostrowski, M.; Mazard, S.; Garczarek, L.; Vaulot, D.; Not, F.; Massana, R.; Ulloa, O.; Scanlan, D.J. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ. Microbiol. 2007, 10, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Ghai, R.; Mizuno, C.M.; Picazo, A.; Camacho, A.; Rodriguez-Valera, F. Metagenomics uncovers a new group of low GC and ultra-small marine Actinobacteria. Sci. Rep. 2013, 3, srep02471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, O.U.; Han, J.; Woyke, T.; Jansson, J.K. Single-cell genomics reveals features of a Colwellia species that was dominant during the Deepwater Horizon oil spill. Front. Microbiol. 2014, 5, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugenholtz, P.; Tyson, G. Metagenomics. Nature 2008, 455, 481–483. [Google Scholar] [CrossRef]
- Sedlar, K.; Kupkova, K.; Provazník, I. Bioinformatics strategies for taxonomy independent binning and visualization of sequences in shotgun metagenomics. Comput. Struct. Biotechnol. J. 2016, 15, 48–55. [Google Scholar] [CrossRef]
- Papudeshi, B.; Haggerty, J.M.; Doane, M.; Morris, M.; Walsh, K.; Beattie, D.T.; Pande, D.; Zaeri, P.; Silva, G.G.Z.; Thompson, F.; et al. Optimizing and evaluating the reconstruction of Metagenome-assembled microbial genomes. BMC Genomics 2017, 18, 915. [Google Scholar] [CrossRef] [Green Version]
- Tully, B.J.; Sachdeva, R.; Graham, E.D.; Heidelberg, J.F. 290 metagenome-assembled genomes from the Mediterranean Sea: A resource for marine microbiology. PeerJ 2017, 5, e3558. [Google Scholar] [CrossRef] [Green Version]
- Stewart, R.D.; Auffret, M.; Warr, A.; Wiser, A.H.; Press, M.O.; Langford, K.W.; Liachko, I.; Snelling, T.J.; Dewhurst, R.J.; Walker, A.W.; et al. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 2018, 9, 870. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, L.G.E.; Ettinger, C.L.; Jospin, G.; Eisen, J.A. Metagenome-assembled genomes provide new insight into the microbial diversity of two thermal pools in Kamchatka, Russia. Sci. Rep. 2019, 9, 3059. [Google Scholar] [CrossRef] [Green Version]
- Voget, S.; Wemheuer, B.; Brinkhoff, T.; Vollmers, J.; Dietrich, S.; Giebel, H.-A.; Beardsley, C.; Sardemann, C.; Bakenhus, I.; Billerbeck, S.; et al. Adaptation of an abundant Roseobacter RCA organism to pelagic systems revealed by genomic and transcriptomic analyses. ISME J. 2014, 9, 371–384. [Google Scholar] [CrossRef] [Green Version]
- Brakstad, O.G.; Throne-Holst, M.; Netzer, R.; Stoeckel, D.; Atlas, R.M. Microbial communities related to biodegradation of dispersed M acondo oil at low seawater temperature with Norwegian coastal seawater. Microb. Biotechnol. 2015, 8, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Tribelli, P.M.; López, N.I. Reporting Key Features in Cold-Adapted Bacteria. Life 2018, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, A.; Syutsubo, K.; Harayama, S. Alcanivorax which prevails in oil-contaminated seawater exhibits broad substrate specificity for alkane degradation. Environ. Microbiol. 2003, 5, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Hara, A.; Baik, S.-H.; Syutsubo, K.; Misawa, N.; Smits, T.H.; Van Beilen, J.B.; Harayama, S. Cloning and functional analysis of alkB genes in Alcanivorax borkumensis SK2. Environ. Microbiol. 2004, 6, 191–197. [Google Scholar] [CrossRef]
- Rizzo, C.; Malavenda, R.; Gerçe, B.; Papale, M.; Syldatk, C.; Hausmann, R.; Bruni, V.; Michaud, L.; Giudice, A.L.; Amalfitano, S. Effects of a Simulated Acute Oil Spillage on Bacterial Communities from Arctic and Antarctic Marine Sediments. Microorganisms 2019, 7, 632. [Google Scholar] [CrossRef] [Green Version]
- Hu, P.; Dubinsky, E.A.; Probst, A.; Wang, J.; Sieber, C.M.K.; Tom, L.M.; Gardinali, P.R.; Banfield, J.; Atlas, R.M.; Andersen, G.L. Simulation of Deepwater Horizonoil plume reveals substrate specialization within a complex community of hydrocarbon degraders. Proc. Natl. Acad. Sci. USA 2017, 114, 7432–7437. [Google Scholar] [CrossRef] [Green Version]
- Dubinsky, E.A.; Conrad, M.E.; Chakraborty, R.; Bill, M.; Borglin, S.E.; Hollibaugh, J.T.; Mason, O.U.; Piceno, Y.M.; Reid, F.C.; Stringfellow, W.T.; et al. Succession of Hydrocarbon-Degrading Bacteria in the Aftermath of the Deepwater Horizon Oil Spill in the Gulf of Mexico. Environ. Sci. Technol. 2013, 47, 10860–10867. [Google Scholar] [CrossRef]
- Vergeynst, L.; Christensen, J.H.; Kjeldsen, K.U.; Meire, L.; Boone, W.; Malmquist, L.M.; Rysgaard, S. In situ biodegradation, photooxidation and dissolution of petroleum compounds in Arctic seawater and sea ice. Water Res. 2018, 148, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Golden, K.M.; Eicken, H.; Heaton, A.L.; Miner, J.; Pringle, D.J.; Zhu, J. Thermal evolution of permeability and microstructure in sea ice. Geophys. Res. Lett. 2007, 34, L16501. [Google Scholar] [CrossRef] [Green Version]
- Faksness, L.-G.; Brandvik, P.J. Distribution of water soluble components from oil encapsulated in Arctic sea ice: Summary of three field seasons. Cold Reg. Sci. Technol. 2008, 54, 106–114. [Google Scholar] [CrossRef]
- Gleitz, M.; Rutgers, V.D.; Loeff, M.; Thomas, D.N.; Dieckmann, G.S.; Millero, F.J. Comparison of summer and winter inorganic carbon, oxygen and nutrient concentrations in Antarctic sea ice brine. Mar. Chem. 1995, 51, 81–91. [Google Scholar] [CrossRef]
- Desmond, D.S.; Saltymakova, D.; Neusitzer, T.D.; Firoozy, D.; Barber, D.G.; Stern, G.A. Oil behavior in sea ice: Changes in chemical composition and resultant effect on sea ice dielectrics. Mar. Pollut. Bull. 2019, 142, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Choo, K.H.; Tong, J.C.; Zhang, L. Recent Applications of Hidden Markov Models in Computational Biology. Genomics Proteom. Bioinform. 2004, 2, 84–96. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, B.R.; Nakatsu, C.H.; Nies, L. Detection and Enumeration of Aromatic Oxygenase Genes by Multiplex and Real-Time PCR. Appl. Environ. Microbiol. 2003, 69, 3350–3358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Yang, J.; Jiang, H.; Zhang, G.; Dong, H. Chemical composition of n-alkanes and microbially mediated n-alkane degradation potential differ in the sediments of Qinghai-Tibetan lakes with different salinity. Chem. Geol. 2019, 524, 37–48. [Google Scholar] [CrossRef]
- Karthikeyan, S.; Hatt, J.K.; Kim, M.; Spain, J.C.; Huettel, M.; Kostka, J.E.; Konstantinidis, K.T. A novel, divergent alkane monooxygenase (alkB) clade involved in crude oil biodegradation. Environ. Microbiol. Rep. 2021, 13, 830–840. [Google Scholar] [CrossRef]
- Brakstad, O.G.; Davies, E.J.; Ribicic, D.; Winkler, A.; Brönner, U.; Netzer, R. Biodegradation of dispersed oil in natural seawaters from Western Greenland and a Norwegian fjord. Polar Biol. 2018, 41, 2435–2450. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Zhang, F.; He, J.; Ji, Z.; Zhou, Q. Water masses influence bacterioplankton community structure in summer Kongsfjorden. Extremophiles 2019, 24, 107–120. [Google Scholar] [CrossRef]
- Guibert, L.M.; Loviso, C.L.; Marcos, M.S.; Commendatore, M.G.; Dionisi, H.M.; Lozada, M. Alkane Biodegradation Genes from Chronically Polluted Subantarctic Coastal Sediments and Their Shifts in Response to Oil Exposure. Microb. Ecol. 2012, 64, 605–616. [Google Scholar] [CrossRef]
- Vollmers, J.; Voget, S.; Dietrich, S.; Gollnow, K.; Smits, M.; Meyer, K.; Brinkhoff, T.; Simon, M.; Daniel, R. Poles Apart: Arctic and Antarctic Octadecabacter strains Share High Genome Plasticity and a New Type of Xanthorhodopsin. PLoS ONE 2013, 8, e63422. [Google Scholar] [CrossRef] [Green Version]
- Zhong, C.; Chen, C.; Wang, L.; Ning, K. Integrating pan-genome with metagenome for microbial community profiling. Comput. Struct. Biotechnol. J. 2021, 19, 1458–1466. [Google Scholar] [CrossRef]
- Qin, W.; Heal, K.R.; Ramdasi, R.; Kobelt, J.N.; Martens-Habbena, W.; Bertagnolli, A.D.; Amin, S.; Walker, C.B.; Urakawa, H.; Koenneke, M.; et al. Nitrosopumilus maritimus gen. nov., sp. nov., Nitrosopumilus cobalaminigenes sp. nov., Nitrosopumilus oxyclinae sp. nov., and Nitrosopumilus ureiphilus sp. nov., four marine ammonia-oxidizing archaea of the phylum Thaumarchaeota. Int. J. Syst. Evol. Microbiol. 2017, 67, 5067–5079. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.-K.; Kim, B.K.; Song, J.Y.; Kwak, M.-J.; Lee, C.H.; Yoon, J.-H.; Oh, T.K.; Kim, J.F. Genomic makeup of the marine flavobacterium Nonlabens (Donghaeana) dokdonensis and identification of a novel class of rhodopsins. Genome Biol. Evol. 2013, 5, 187–199. [Google Scholar] [CrossRef] [Green Version]
- Sampaio, D.S.; Almeida, J.R.B.; De Jesus, H.E.; Rosado, A.S.; Seldin, L.; Jurelevicius, D. Distribution of anaerobic hydrocar-bon-degrading bacteria in soils from King George Island, Maritime Antarctica. Microb. Ecol. 2017, 74, 810–820. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peeb, A.; Dang, N.P.; Truu, M.; Nõlvak, H.; Petrich, C.; Truu, J. Assessment of Hydrocarbon Degradation Potential in Microbial Communities in Arctic Sea Ice. Microorganisms 2022, 10, 328. https://doi.org/10.3390/microorganisms10020328
Peeb A, Dang NP, Truu M, Nõlvak H, Petrich C, Truu J. Assessment of Hydrocarbon Degradation Potential in Microbial Communities in Arctic Sea Ice. Microorganisms. 2022; 10(2):328. https://doi.org/10.3390/microorganisms10020328
Chicago/Turabian StylePeeb, Angela, Nga Phuong Dang, Marika Truu, Hiie Nõlvak, Chris Petrich, and Jaak Truu. 2022. "Assessment of Hydrocarbon Degradation Potential in Microbial Communities in Arctic Sea Ice" Microorganisms 10, no. 2: 328. https://doi.org/10.3390/microorganisms10020328
APA StylePeeb, A., Dang, N. P., Truu, M., Nõlvak, H., Petrich, C., & Truu, J. (2022). Assessment of Hydrocarbon Degradation Potential in Microbial Communities in Arctic Sea Ice. Microorganisms, 10(2), 328. https://doi.org/10.3390/microorganisms10020328