
Parameterising Translational Feedback Models of Autoregulatory RNA-Binding Proteins in Saccharomyces cerevisiae


Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection of Potential Autoregulatory RBPs
2.2. Data Acquisition
2.3. Acquisition and Conversion of Single-Cell Association/Dissociation Constants
3. Results
3.1. Modelling RBP Gene Expression without Feedback Is a Poor Predictor of the Steady State Protein Abundance
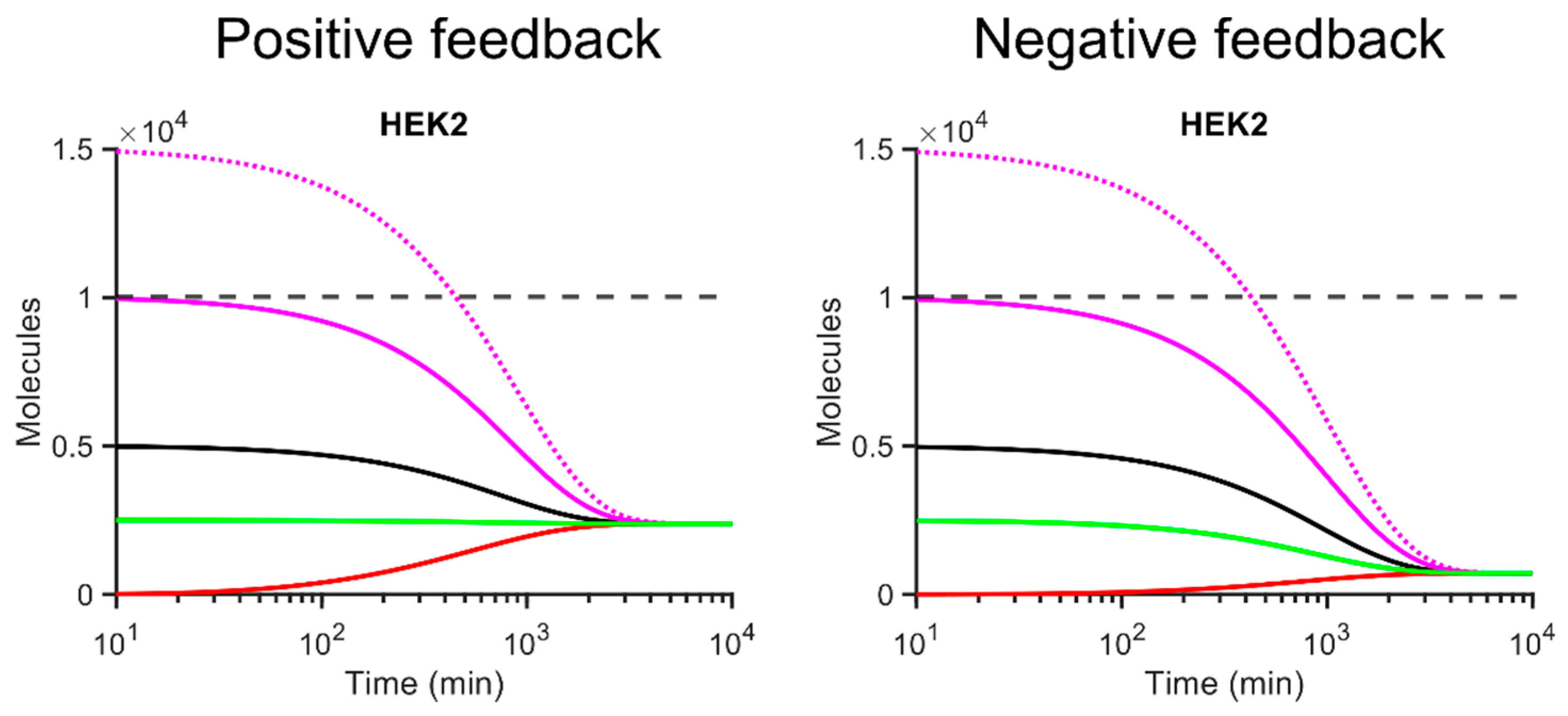
3.2. Gene Expression and Steady State Predictions Can Be Tuned Using Translational Feedback Loops
3.3. Co-Operative Positive Translational Feedback Simulations Can Predict More Accurate Steady State Protein Abundances Than Models without Feedback
3.4. Negative Translational Feedback Is Highly Repressive at Biologically Relevant Scales
3.5. Pfk2p and Map1p Protein Levels Are Better Predicted with Positive or Negative Feedback Loops Compared to Models without Feedback
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merrick, W.C. eIF4F: A retrospective. J. Biol. Chem. 2015, 290, 24091–24099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prévôt, D.; Darlix, J.-L.; Ohlmann, T. Conducting the initiation of protein synthesis: The role of eIF4G. Biol. Cell 2003, 95, 141–156. [Google Scholar] [CrossRef]
- Amorim, I.; Lach, G.; Gkogkas, C. The Role of the Eukaryotic Translation Initiation Factor 4E (eIF4E) in Neuropsychiatric Disorders. Front. Genet. 2018, 9, 561. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Rogers, G.W.; Komar, A.A.; Merrick, W.C. eIF4A: The godfather of the DEAD box helicases. Prog. Nucleic Acid Res. Mol. Biol. 2002, 72, 307–331. [Google Scholar]
- Kahvejian, A.; Svitkin, Y.V.; Sukarieh, R.; M’Boutchou, M.N.; Sonenberg, N. Mammalian poly(A)-binding protein is a eukaryotic translation initiation factor, which acts via multiple mechanisms. Genes Dev. 2005, 19, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Gerber, A.P.; Herschlag, D.; Brown, P.O. Extensive Association of Functionally and Cytotopically Related mRNAs with Puf Family RNA-Binding Proteins in Yeast. PLoS Biol. 2004, 2, e79. [Google Scholar] [CrossRef]
- Keene, J.D. RNA regulons: Coordination of post-transcriptional events. Nat. Rev. Genet. 2007, 8, 533–543. [Google Scholar] [CrossRef]
- Imig, J.; Kanitz, A.; Gerber, A.P. RNA regulons and the RNA-protein interaction network. Biomol. Concepts 2012, 3, 403–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Romano, M.C.; Davidson, F.A. Translational control of gene expression via interacting feedback loops. Phys. Rev. E 2019, 100, 050402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wulund, L.; Reddy, A.B. A brief history of circadian time: The emergence of redox oscillations as a novel component of biological rhythms. Perspect. Sci. 2015, 6, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Barberis, M.; Todd, R.G.; van der Zee, L. Advances and challenges in logical modeling of cell cycle regulation: Perspective for multi-scale, integrative yeast cell models. FEMS Yeast Res. 2017, 17, fow103. [Google Scholar] [CrossRef] [PubMed]
- Leite, M.C.A.; Wang, Y. Multistability, oscillations and bifurcations in feedback loops. Math. Biosci. Eng. 2010, 7, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Müller-McNicoll, M.; Rossbach, O.; Hui, J.; Medenbach, J. Auto-regulatory feedback by RNA-binding proteins. J. Mol. Cell Biol. 2019, 11, 930–939. [Google Scholar] [CrossRef] [Green Version]
- Huh, K.; Falvo, J.V.; Gerke, L.C.; Carroll, A.S.; Howson, R.W.; Weissman, J.S.; O’Shea, E.K. Global analysis of protein localization in budding yeast. Nature 2003, 425, 686–691. [Google Scholar] [CrossRef]
- Roy, B.; Granas, D.; Bragg, F.; Cher, J.A.Y.; White, M.A.; Stormo, G.D. Autoregulation of yeast ribosomal proteins discovered by efficient search for feedback regulation. Commun. Biol. 2020, 3, 761. [Google Scholar] [CrossRef]
- Frugier, M.; Ryckelynck, M.; Giegé, R. tRNA-balanced expression of a eukaryal aminoacyl-tRNA synthetase by an mRNA-mediated pathway. EMBO Rep. 2005, 6, 860–865. [Google Scholar] [CrossRef]
- Levi, O.; Arava, Y. mRNA association by aminoacyl tRNA synthetase occurs at a putative anticodon mimic and autoregulates translation in response to tRNA levels. PLoS Biol. 2019, 17, e3000274. [Google Scholar] [CrossRef]
- Mittal, N.; Scherrer, T.; Gerber, A.P.; Janga, S.C. Interplay between posttranscriptional and posttranslational interactions of RNA-binding proteins. J. Mol. Biol. 2011, 409, 466–479. [Google Scholar] [CrossRef] [Green Version]
- Tyng, V.; Kellman, M.E. Kinetic Model of Translational Autoregulation. J. Phys. Chem. B 2019, 123, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.; Baryshnikova, A.; Brown, G.W. Unification of Protein Abundance Datasets Yields a Quantitative Saccharomyces cerevisiae Proteome. Cell Syst. 2018, 6, 192–205.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackmann, A.; Wu, H.; Schneider, U.-M.; Meyer, K.; Jung, K.; Krebber, H. Quality control of spliced mRNAs requires the shuttling SR proteins Gbp2 and Hrb1. Nat. Commun. 2014, 5, 3123. [Google Scholar] [CrossRef] [PubMed]
- Paquin, N.; Ménade, M.; Poirier, G.; Donato, D.; Drouet, E.; Chartrand, P. Local Activation of Yeast ASH1 mRNA Translation through Phosphorylation of Khd1p by the Casein Kinase Yck1p. Mol. Cell 2007, 26, 795–809. [Google Scholar] [CrossRef]
- Wang, L.; Lewis, M.S.; Johnson, A.W. Domain interactions within the Ski2/3/8 complex and between the Ski complex and Ski7p. RNA 2005, 11, 1291–1302. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, W.; Siebel, C.W.; Guthrie, C. Phosphorylation by Sky1p promotes Npl3p shuttling and mRNA dissociation. RNA 2001, 7, 302–313. [Google Scholar] [CrossRef] [Green Version]
- Moehle, E.A.; Ryan, C.J.; Krogan, N.J.; Kress, T.L.; Guthrie, C. The Yeast SR-Like Protein Npl3 Links Chromatin Modification to mRNA Processing. PLoS Genet. 2012, 8, e1003101. [Google Scholar] [CrossRef] [Green Version]
- Newcomb, L.L.; Diderich, J.A.; Slattery, M.G.; Heideman, W. Glucose Regulation of Saccharomyces cerevisiae Cell Cycle Genes. Eukaryot. Cell 2003, 2, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Chang, Y.H. Amino-terminal protein processing in Saccharomyces cerevisiae is an essential function that requires two distinct methionine aminopeptidases. Proc. Natl. Acad. Sci. USA 1995, 92, 12357–12361. [Google Scholar] [CrossRef] [Green Version]
- Scherrer, T.; Mittal, N.; Janga, S.C.; Gerber, A.P. A Screen for RNA-Binding Proteins in Yeast Indicates Dual Functions for Many Enzymes. PLoS ONE 2010, 5, e15499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matia-González, A.M.; Laing, E.E.; Gerber, A.P. Conserved mRNA-binding proteomes in eukaryotic organisms. Nat. Struct. Mol. Biol. 2015, 22, 1027–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelechano, V.; Chávez, S.; Pérez-Ortín, J.E. A Complete Set of Nascent Transcription Rates for Yeast Genes. PLoS ONE 2010, 5, e15442. [Google Scholar] [CrossRef] [PubMed]
- Brachmann, C.B.; Davies, A.; Cost, G.J.; Caputo, E. Designer Deletion Strains derived from Saccharomyces cerevisiae S288C: A Useful set of Strains and Plasmids for PCR-mediated Gene Disruption and Other Applications. Yeast 1998, 14, 115–132. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, C.L.; Storey, J.D.; Tibshirani, R.J.; Herschlag, D.; Brown, P.O. Precision and functional specificity in mRNA decay. Proc. Natl. Acad. Sci. USA 2002, 99, 5860–5865. [Google Scholar] [CrossRef] [Green Version]
- Nonet, M.; Scafe, C.; Sexton, J.; Young, R. Eucaryotic RNA Polymerase Conditional Mutant That Rapidly Ceases mRNA Synthesis. Mol. Cell. Biol. 1987, 7, 1602–1611. [Google Scholar]
- Riba, A.; Di Nanni, N.; Mittal, N.; Arhné, E.; Schmidt, A.; Zavolan, M. Protein synthesis rates and ribosome occupancies reveal determinants of translation elongation rates. Proc. Natl. Acad. Sci. USA 2019, 116, 15023–15032. [Google Scholar] [CrossRef] [Green Version]
- Belle, A.; Tanay, A.; Bitincka, L.; Shamir, R.; O’Shea, E.K. Quantification of protein half-lives in the budding yeast proteome. Proc. Natl. Acad. Sci. USA 2006, 103, 13004–13009. [Google Scholar] [CrossRef] [Green Version]
- Ghaemmaghami, S.; Huh, W.-K.; Bower, K.; Howson, R.W.; Belle, A.; Dephoure, N.; O’Shea, E.K.; Weissman, J.S. Global analysis of protein expression in yeast. Nature 2003, 425, 737–741. [Google Scholar] [CrossRef]
- Miura, F.; Kawaguchi, N.; Yoshida, M.; Uematsu, C.; Kito, K.; Sakaki, Y.; Ito, T. Absolute quantification of the budding yeast transcriptome by means of competitive PCR between genomic and complementary DNAs. BMC Genom. 2008, 9, 574. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Lumbreras, S.; Taverniti, V.; Zorrilla, S.; Séraphin, B.; Pérez-Cañadillas, J.M. Gbp2 interacts with THO/TREX through a novel type of RRM domain. Nucleic Acids Res. 2016, 44, 437–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, Y.; Irie, K.; Gerber, A.P. Distinct roles for Khd1p in the localization and expression of bud-localized mRNAs in yeast. RNA 2008, 14, 2333–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halbach, F.; Rode, M.; Conti, E. The crystal structure of S. cerevisiae Ski2, a DExH helicase associated with the cytoplasmic functions of the exosome. RNA 2012, 18, 124–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deka, P.; Bucheli, M.E.; Moore, C.; Buratowski, S.; Varani, G. Structure of the Yeast SR Protein Npl3 and Interaction with mRNA 3′-End Processing Signals. J. Mol. Biol. 2008, 375, 136–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, P.; Nishikawa, J.L.; Breitkreutz, B.J.; Tyers, M. Systematic identification of pathways that couple cell growth and division in yeast. Science 2002, 297, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Greenhill, E.R.; Rocco, A.; Vibert, L.; Nikaido, M.; Kelsh, R.N. An Iterative Genetic and Dynamical Modelling Approach Identifies Novel Features of the Gene Regulatory Network Underlying Melanocyte Development. PLoS Genet. 2011, 7, e1002265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elowitz, M.B.; Leibier, S. A synthetic oscillatory network of transcriptional regulators. Nature 2000, 403, 335–338. [Google Scholar] [CrossRef]
- Williams, J.W.; Cui, X.; Levchenko, A.; Stevens, A.M. Robust and sensitive control of a quorum-sensing circuit by two interlocked feedback loops. Mol. Syst. Biol. 2008, 4, 234. [Google Scholar] [CrossRef]
- Farjami, S.; Sosa, K.C.; Dawes, J.H.P.; Kelsh, R.N.; Rocco, A. Novel generic models for differentiating stem cells reveal oscillatory mechanisms. J. R. Soc. Interface 2021, 18, 20210442. [Google Scholar] [CrossRef]
- Dublanche, Y.; Michalodimitrakis, K.; Kü, N.; Foglierini, M.; Serrano, L. Noise in transcription negative feedback loops: Simulation and experimental analysis. Mol. Syst. Biol. 2006, 2, 41. [Google Scholar] [CrossRef] [Green Version]
- Hornung, G.; Barkai, N. Noise Propagation and Signaling Sensitivity in Biological Networks: A Role for Positive Feedback. PLoS Comput. Biol. 2008, 4, e8. [Google Scholar] [CrossRef] [Green Version]
- Hurley, J.M.; Loros, J.J.; Dunlap, J.C.; Hurley, J.M.; Dunlap, J.C. Circadian Oscillators: Around the Transcription-Translation Feedback Loop and on to Output. Trends Biochem. Sci. 2016, 41, 834–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Jacobs-Wagner, C. Effects of mRNA Degradation and Site-Specific Transcriptional Pausing on Protein Expression Noise. Biophys. J. 2018, 114, 1718–1729. [Google Scholar] [CrossRef] [PubMed]
- Aquino, G.; Rocco, A. Bimodality in gene expression without feedback: From Gaussian white noise to log-normal coloured noise. Math. Biosci. Eng. 2020, 17, 6993–7071. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.R.S.; Ghaemmaghami, S.; Ihmels, J.; Breslow, D.K.; Noble, M.; Derisi, J.L.; Weissman, J.S. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature 2006, 441, 840–846. [Google Scholar] [CrossRef]
- Castells-Roca, L.; García-Martínez, J.; Moreno, J.; Herrero, E.; Bellí, G. Heat Shock Response in Yeast Involves Changes in Both Transcription Rates and mRNA Stabilities. PLoS ONE 2011, 6, e17272. [Google Scholar] [CrossRef] [Green Version]
- Verghese, J.; Abrams, J.; Wang, Y.; Morano, K.A. Biology of the Heat Shock Response and Protein Chaperones: Budding Yeast (Saccharomyces cerevisiae) as a Model System. Microbiol. Mol. Biol. Rev. 2012, 76, 115–158. [Google Scholar] [CrossRef] [Green Version]
- Schmiedel, J.M.; Klemm, S.K.; Zheng, Y.; Sahay, A.; Blüthgen, N.; Marks, D.S.; van Oudenaarden, A. MicroRNA control of protein expression noise. Science 2015, 348, 128–132. [Google Scholar] [CrossRef]
- Cech, T.R. The RNA worlds in context. Cold Spring Harb. Perspect. Biol. 2012, 4, a006742. [Google Scholar] [CrossRef] [Green Version]
- Espah Borujeni, A.; Salis, H.M. Translation Initiation is Controlled by RNA Folding Kinetics via a Ribosome Drafting Mechanism. J. Am. Chem. Soc. 2016, 138, 7016–7023. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | HRB1 | HEK2 | SKI2 | NPL3 | PFK2 | MAP1 |
|---|---|---|---|---|---|---|
| k1 | 1.08 × 10−1 | 1.97 × 10−1 | 1.35 × 10−1 | 3.26 × 10−1 | 5.79 × 10−1 | 4.90 × 10−1 |
| k2 | 7.82 × 10−2 | 8.19 × 10−2 | 3.01 × 10−2 | 2.46 × 10−2 | 2.26 × 10−2 | 3.70 × 10−2 |
| k3 | 1.13 | 3.99 × 10−1 | 4.97 × 10−1 | 9.23 | 1.54 × 10 | 1.03 × 10 |
| k4 | 6.10 × 10−4 | 9.66 × 10−4 | 1.23 × 10−3 | 1.16 × 10−3 | 1.06 × 10−3 | 1.55 × 10−3 |
| Pss | 4.79 × 103 | 1.00 × 104 | 4.62 × 103 | 4.78 × 104 | 9.44 × 104 | 1.52 × 104 |
| mss | 3.00 | 8.55 | 1.37 × 10 | 5.10 × 10−1 | 2.15 × 10 | 1.15 × 10 |
| Kd | 6.05 × 104 | 1.8 × 103 | 1.01 × 104 | 5.04 × 10 | 2.17 × 105 * | 6.14 × 104 * |
| Ka | 1.65 × 10−5 1.65 × 10−5 * | 5.56 × 10−4 | 9.90 × 10−5 | 1.98 × 10−2 2.60 × 10−5 * | 2.43 × 10−5 * | 2.67 × 10−4 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clarke-Whittet, M.; Rocco, A.; Gerber, A.P. Parameterising Translational Feedback Models of Autoregulatory RNA-Binding Proteins in Saccharomyces cerevisiae. Microorganisms 2022, 10, 340. https://doi.org/10.3390/microorganisms10020340
Clarke-Whittet M, Rocco A, Gerber AP. Parameterising Translational Feedback Models of Autoregulatory RNA-Binding Proteins in Saccharomyces cerevisiae. Microorganisms. 2022; 10(2):340. https://doi.org/10.3390/microorganisms10020340
Chicago/Turabian StyleClarke-Whittet, Michael, Andrea Rocco, and André P. Gerber. 2022. "Parameterising Translational Feedback Models of Autoregulatory RNA-Binding Proteins in Saccharomyces cerevisiae" Microorganisms 10, no. 2: 340. https://doi.org/10.3390/microorganisms10020340