
Translational Fusion to Hmp Improves Heterologous Protein Expression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Plasmids
2.2. Growth Media and Chemicals
2.3. Fluorophore Expression Assay
2.4. β-Galactosidase Assay
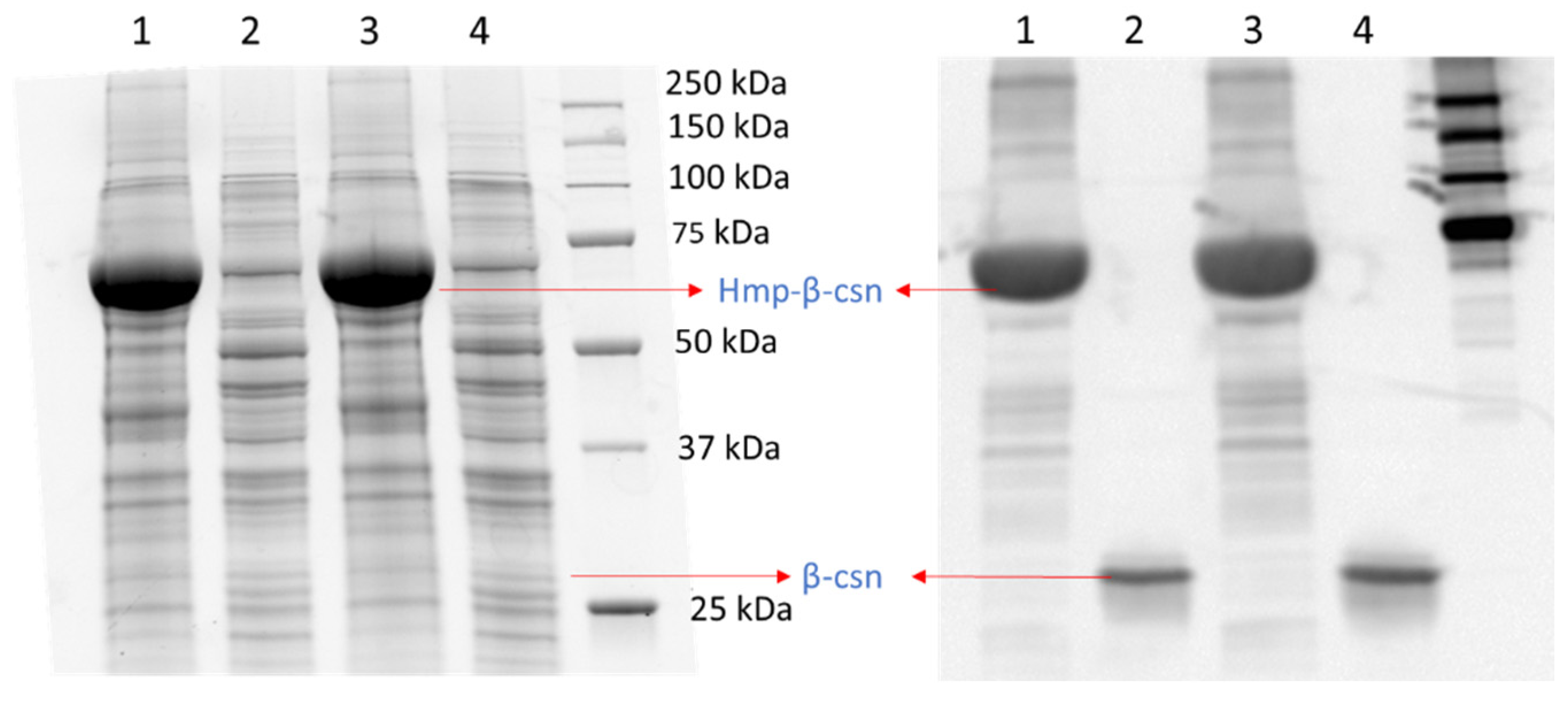
2.5. Detection of LacZ, sfGFP, and Their Hmp Fusions by SDS-PAGE
2.6. β-Casein Expression and Detection by Western Blot
2.7. Protein Degradation Assay
2.8. qPCR
2.9. Protein Solubility Assay
2.10. Fusion Tag Cleavage Assay
2.11. RBS Calculator
3. Results
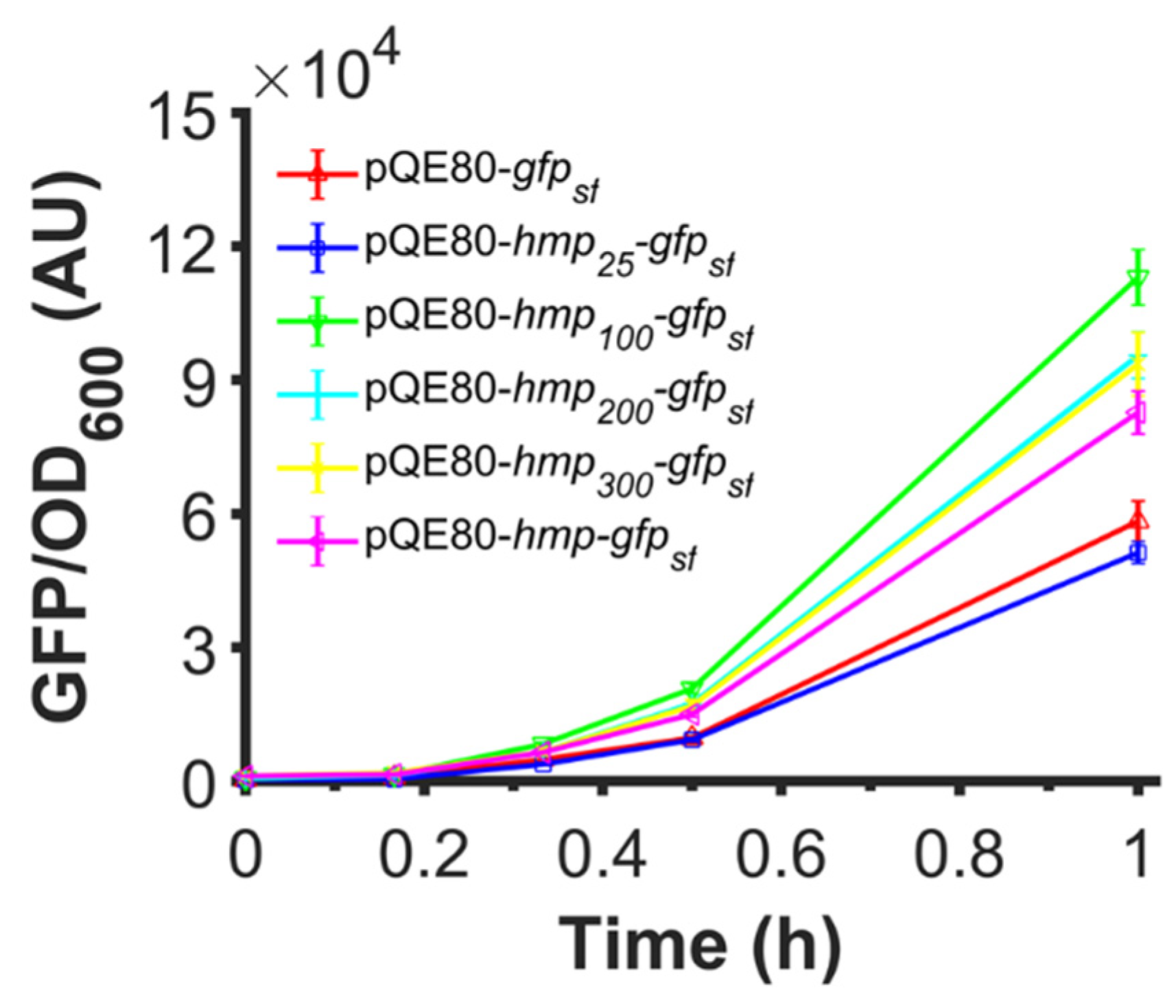
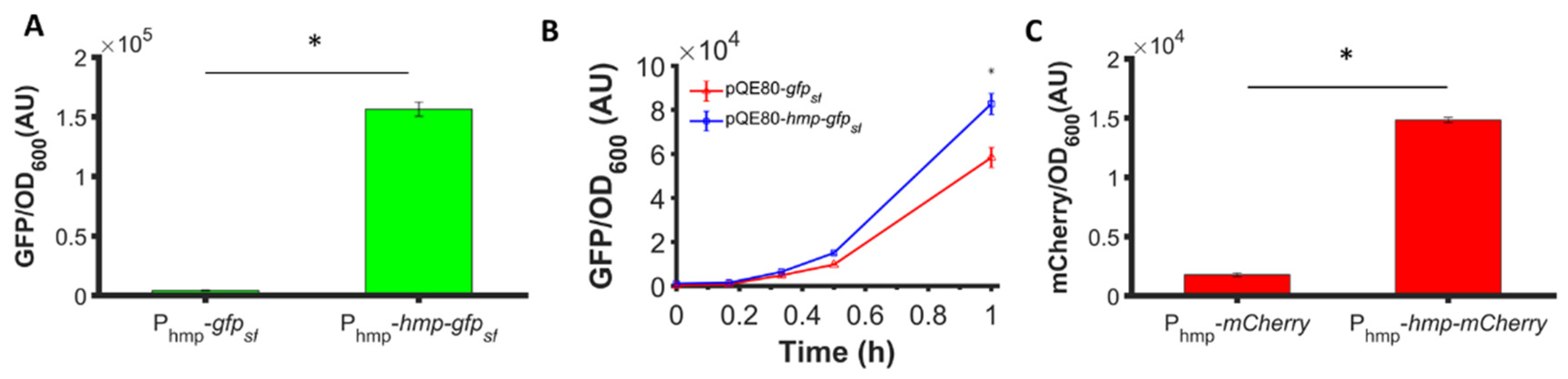
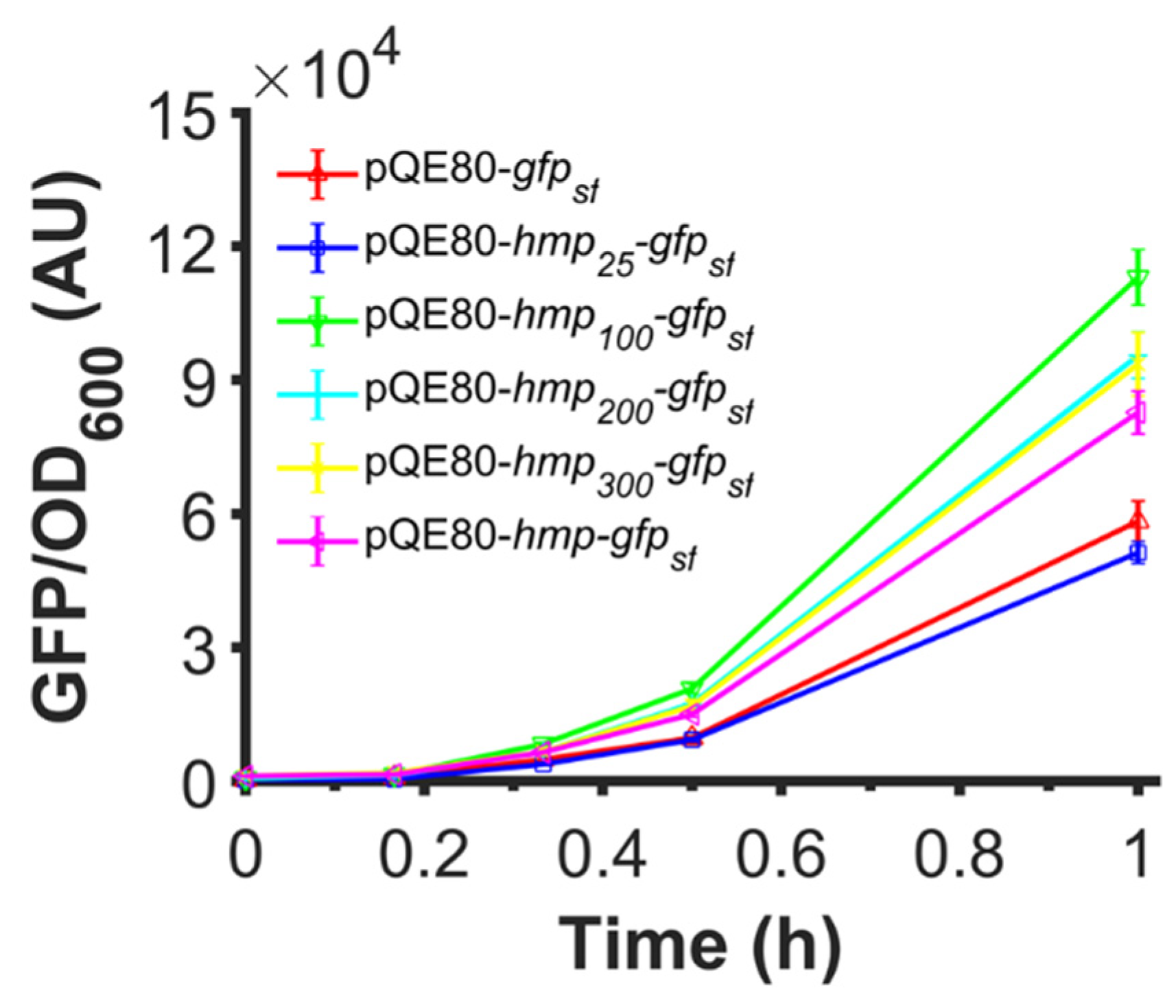
3.1. Fusion of Hmp to sfGFP Increases Expression
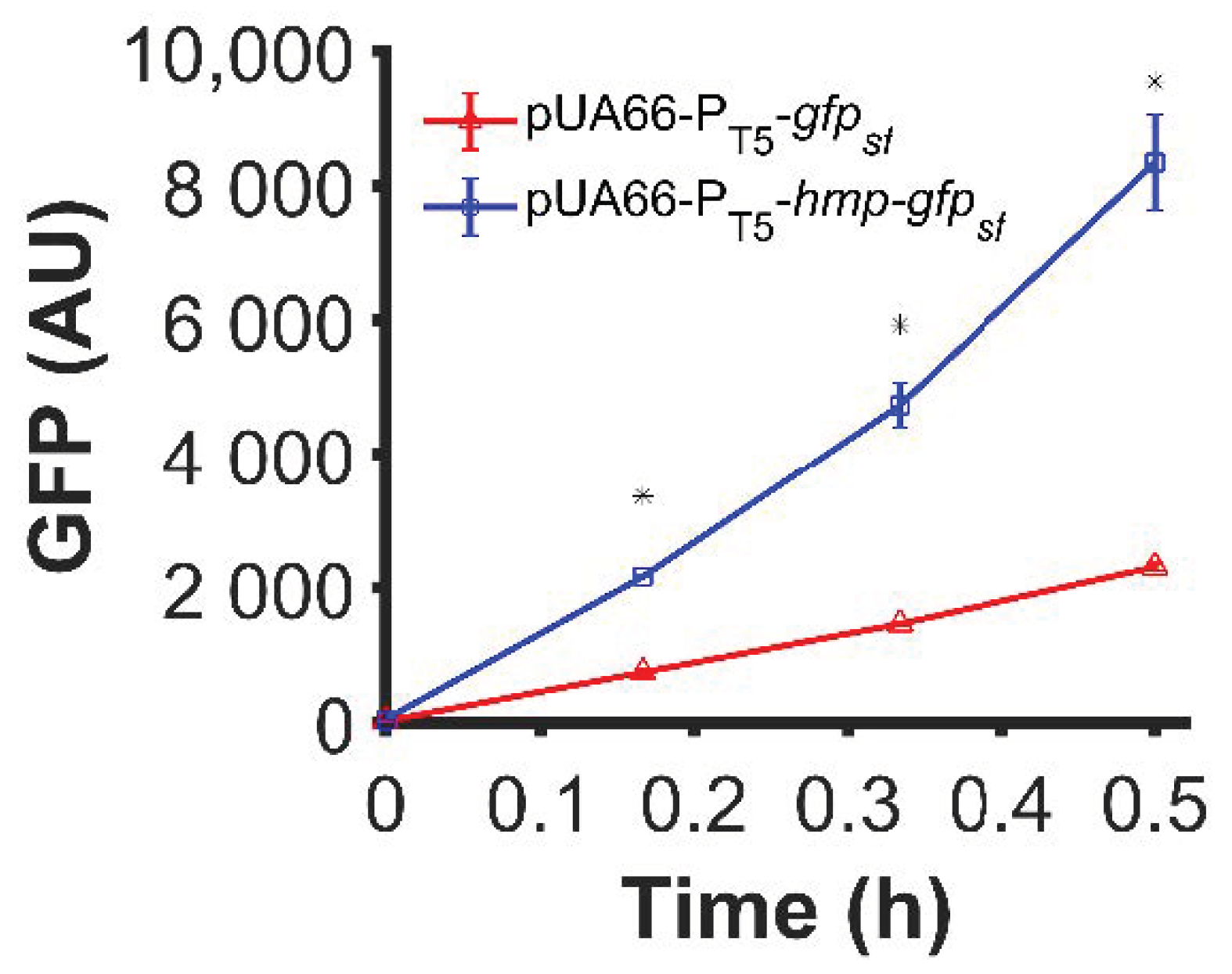
3.2. Fusion to Hmp Increases the Expression of Different Proteins from Different Promoters in Different Plasmids and Different Media
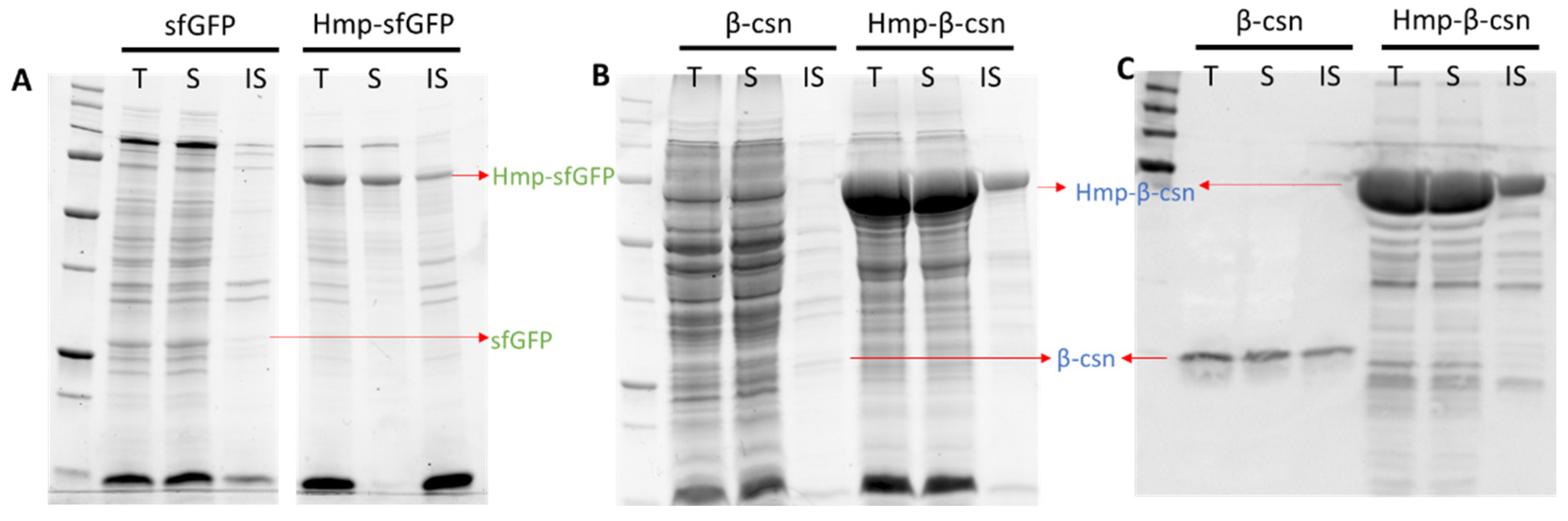
3.3. Hmp Fusion Proteins Are Largely in the Soluble Fraction of Lysates
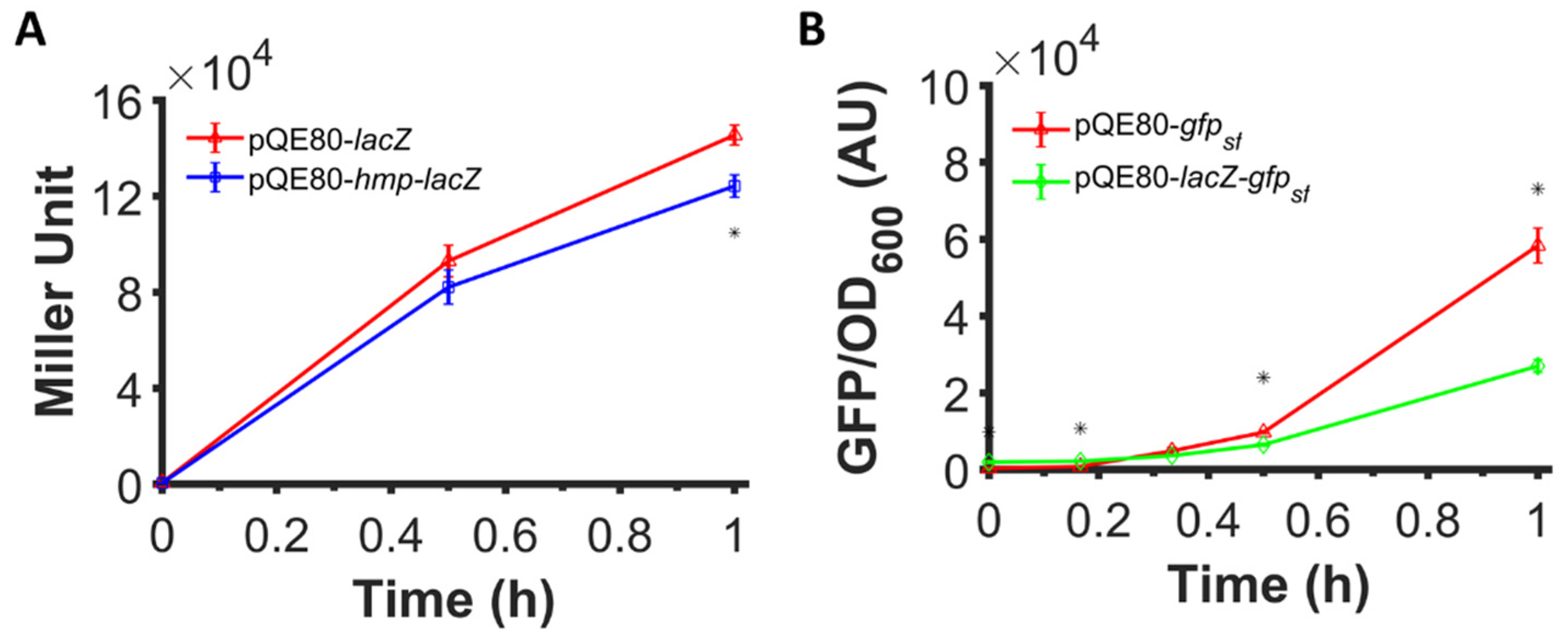
3.4. Fusion of Hmp to LacZ Does Not Increase Expression
3.5. Catalytic Activity Is Not Required to Observe Expression Enhancement from Hmp Fusions
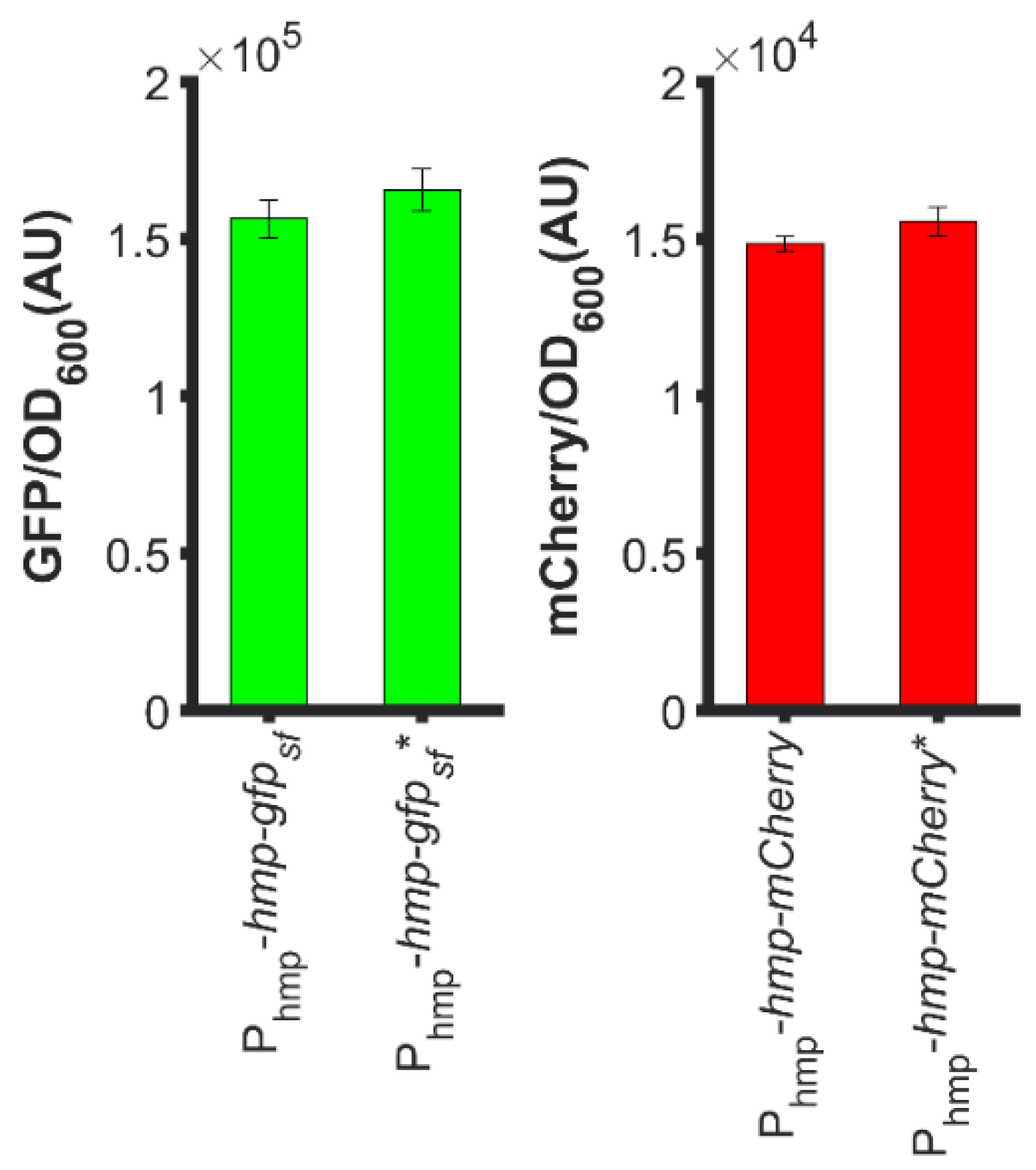
3.6. Fusion of Hmp to the N-Terminus of Proteins Is Required for Increased Expression
3.7. Increased Expression Is Not Due to a Second Start Codon
3.8. RBS Calculator Predicts the Impact of an N-Terminal Linker but Not the Increased Expression of Hmp Fusions
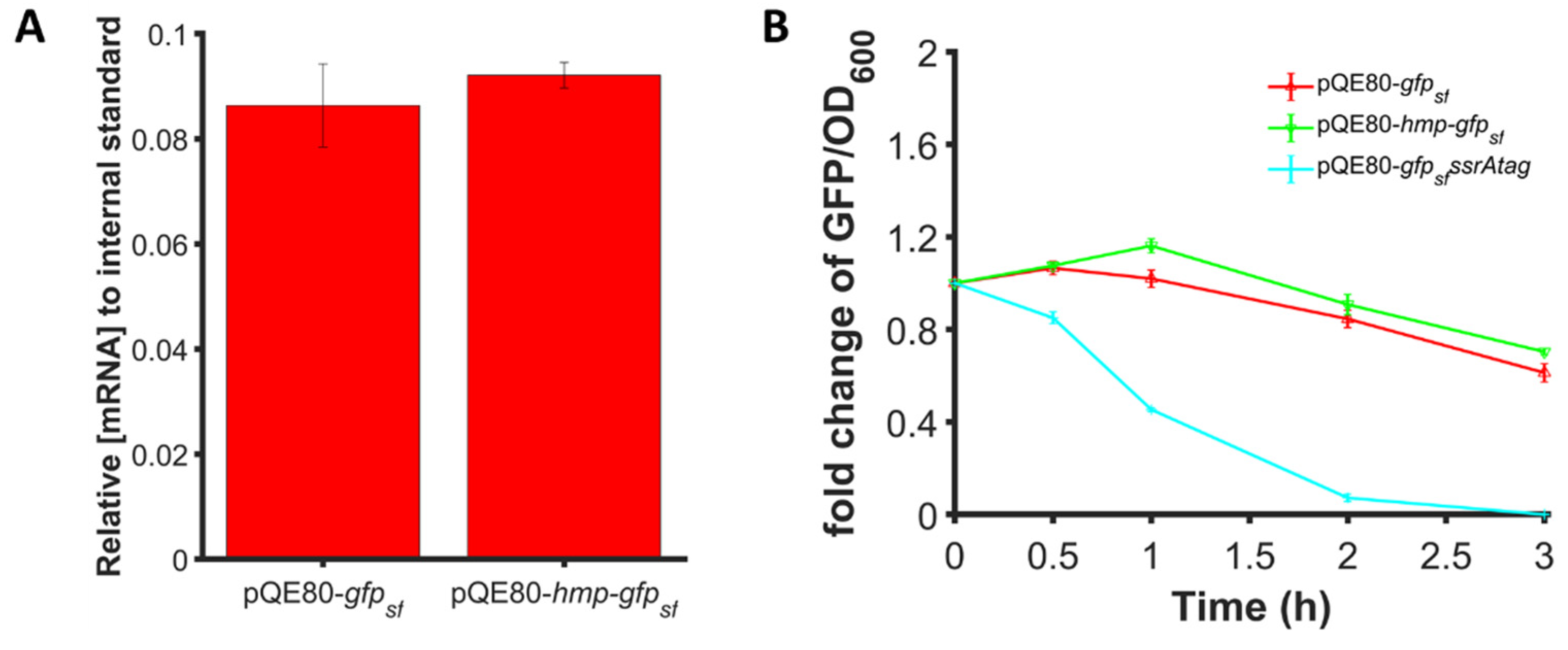
3.9. Transcript Levels and Protein Degradation Are Comparable between Hmp-sfGFP and sfGFP
3.10. Fusions of Truncated Versions of Hmp Also Increase Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Gardner, P.R.; Gardner, A.M.; Martin, L.A.; Salzman, A.L. Nitric oxide dioxygenase: An enzymic function for flavohemoglobin. Proc. Natl. Acad. Sci. USA 1998, 95, 10378. [Google Scholar] [CrossRef] [Green Version]
- Bonamore, A.; Boffi, A. Flavohemoglobin: Structure and reactivity. IUBMB Life 2008, 60, 19–28. [Google Scholar] [CrossRef]
- Hernández-Urzúa, E.; Mills, C.E.; White, G.P.; Contreras-Zentella, M.L.; Escamilla, E.; Vasudevan, S.G.; Membrillo-Hernández, J.; Poole, R.K. Flavohemoglobin Hmp, but Not Its Individual Domains, Confers Protection from Respiratory Inhibition by Nitric Oxide in Escherichia coli. J. Biol. Chem. 2003, 278, 34975–34982. [Google Scholar] [CrossRef] [Green Version]
- Gardner, A.M.; Martin, L.A.; Gardner, P.R.; Dou, Y.; Olson, J.S. Steady-state and transient kinetics of Escherichia coli nitric-oxide dioxygenase (flavohemoglobin). The B10 tyrosine hydroxyl is essential for dioxygen binding and catalysis. J. Biol. Chem. 2000, 275, 12581–12589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.L.; Brynildsen, M.P. Discovery and dissection of metabolic oscillations in the microaerobic nitric oxide response network of Escherichia coli. Proc. Natl. Acad. Sci. USA 2016, 113, E1757–E1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.O.; Orii, Y.; Lloyd, D.; Hughes, M.N.; Poole, R.K. Anoxic function for the Escherichia coli flavohaemoglobin (Hmp): Reversible binding of nitric oxide and reduction to nitrous oxide. FEBS Lett. 1999, 445, 389–394. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Corker, H.; Orii, Y.; Poole, R.K. Escherichia coli Hmp, an “oxygen-binding flavohaemoprotein”, produces superoxide anion and self-destructs. Arch. Microbiol. 2004, 182, 193–203. [Google Scholar] [CrossRef]
- Bodenmiller Diane, M.; Spiro, S. The yjeB (nsrR) Gene of Escherichia coli Encodes a Nitric Oxide-Sensitive Transcriptional Regulator. J. Bacteriol. 2006, 188, 874–881. [Google Scholar] [CrossRef] [Green Version]
- Tucker, N.P.; Le Brun, N.E.; Dixon, R.; Hutchings, M.I. There′s NO stopping NsrR, a global regulator of the bacterial NO stress response. Trends Microbiol. 2010, 18, 149–156. [Google Scholar] [CrossRef]
- Poole, R.K. Flavohaemoglobin: The pre-eminent nitric oxide-detoxifying machine of microorganisms. F1000Research 2020, 9, F1000. [Google Scholar] [CrossRef] [Green Version]
- Khosla, C.; Bailey, J.E. Heterologous expression of a bacterial haemoglobin improves the growth properties of recombinant Escherichia coli. Nature 1988, 331, 633–635. [Google Scholar] [CrossRef] [PubMed]
- Khosla, C.; Curtis, J.E.; Bydalek, P.; Swartz, J.R.; Bailey, J.E. Expression of Recombinant Proteins in Escherichia coli Using an Oxygen-Responsive Promoter. Bio/Technology 1990, 8, 554–558. [Google Scholar] [CrossRef]
- Arnaldos, M.; Kunkel, S.A.; Wang, J.; Pagilla, K.R.; Stark, B.C. Vitreoscilla hemoglobin enhances ethanol production by Escherichia coli in a variety of growth media. Biomass Bioenergy 2012, 37, 1–8. [Google Scholar] [CrossRef]
- Pablos, T.E.; Mora, E.M.; Le Borgne, S.; Ramírez, O.T.; Gosset, G.; Lara, A.R. Vitreoscilla hemoglobin expression in engineered Escherichia coli: Improved performance in high cell-density batch cultivations. Biotechnol. J. 2011, 6, 993–1002. [Google Scholar] [CrossRef]
- Kallio, P.T.; Bailey, J.E. Intracellular expression of Vitreoscilla hemoglobin (VHb) enhances total protein secretion and improves the production of α-amylase and neutral protease in Bacillus subtilis. Biotechnol. Prog. 1996, 12, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Kahraman, H.; Erenler, S.O. Rhamnolipid production by Pseudomonas aeruginosa engineered with the Vitreoscilla hemoglobin gene. Appl. Biochem. Microbiol. 2012, 48, 188–193. [Google Scholar] [CrossRef]
- Wang, T.; Bai, L.; Zhu, D.; Lei, X.; Liu, G.; Deng, Z.; You, D. Enhancing macrolide production in Streptomyces by coexpressing three heterologous genes. Enzym. Microb. Technol. 2012, 50, 5–9. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y. Modulating betulinic acid production in Saccharomyces cerevisiae by managing the intracellular supplies of the co-factor NADPH and oxygen. J. Biosci. Bioeng. 2015, 119, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-M.; Fu, W.-C. Intracellular co-expression of Vitreoscilla hemoglobin enhances cell performance and β-galactosidase production in Pichia pastoris. J. Biosci. Bioeng. 2012, 113, 332–337. [Google Scholar] [CrossRef]
- Wen, Y.; Song, Y.; Li, J.L. [The effects of Vitreoscilla hemoglobin expression on growth and antibiotic production in Streptomyces cinnamonensis]. Sheng Wu Gong Cheng Xue Bao = Chin. J. Biotechnol. 2001, 17, 24–28. [Google Scholar]
- Frey, A.D.; Kallio, P.T. Nitric oxide detoxification—A new era for bacterial globins in biotechnology? Trends Biotechnol. 2005, 23, 69–73. [Google Scholar] [CrossRef]
- Wei, X.-X.; Chen, G.-Q. Chapter Fifteen—Applications of the VHb Gene vgb for Improved Microbial Fermentation Processes. In Methods in Enzymology; Poole, R.K., Ed.; Academic Press: Cambridge, MA, USA, 2008; Volume 436, pp. 273–287. [Google Scholar]
- Kallio, P.T.; Kim, D.J.; Tsai, P.S.; Bailey, J.E. Intracellular expression of Vitreoscilla hemoglobin alters Escherichia coli energy metabolism under oxygen-limited conditions. Eur. J. Biochem. 1994, 219, 201–208. [Google Scholar] [CrossRef]
- Kaur, R.; Pathania, R.; Sharma, V.; Mande Shekhar, C.; Dikshit Kanak, L. Chimeric Vitreoscilla Hemoglobin (VHb) Carrying a Flavoreductase Domain Relieves Nitrosative Stress in Escherichia coli: New Insight into the Functional Role of VHb. Appl. Environ. Microbiol. 2002, 68, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Park, K.-W.; Kim, K.-J.; Howard, A.J.; Stark, B.C.; Webster, D.A. Vitreoscilla Hemoglobin Binds to Subunit I of Cytochrome bo Ubiquinol Oxidases. J. Biol. Chem. 2002, 277, 33334–33337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, A.D.; Shepherd, M.; Jokipii-Lukkari, S.; Häggman, H.; Kallio, P.T. Chapter 3—The Single-Domain Globin of Vitreoscilla: Augmentation of Aerobic Metabolism for Biotechnological Applications. In Advances in Microbial Physiology; Poole, R.K., Ed.; Academic Press: Cambridge, MA, USA, 2011; Volume 58, pp. 81–139. [Google Scholar]
- Webster, D.A.; Dikshit, K.L.; Pagilla, K.R.; Stark, B.C. The Discovery of Vitreoscilla Hemoglobin and Early Studies on Its Biochemical Functions, the Control of Its Expression, and Its Use in Practical Applications. Microorganisms 2021, 9, 1637. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Brynildsen, M.P. Robustness of nitric oxide detoxification to nitrogen starvation in Escherichia coli requires RelA. Free Radic. Biol. Med. 2021, 176, 286–297. [Google Scholar] [CrossRef]
- Robinson, J.L.; Brynildsen, M.P. An ensemble-guided approach identifies ClpP as a major regulator of transcript levels in nitric oxide-stressed Escherichia coli. Metab. Eng. 2015, 31, 22–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2006, 2, 2006.0008. [Google Scholar] [CrossRef] [Green Version]
- Orman, M.A.; Brynildsen, M.P. Dormancy Is Not Necessary or Sufficient for Bacterial Persistence. Antimicrob. Agents Chemother. 2013, 57, 3230–3239. [Google Scholar] [CrossRef] [Green Version]
- Zaslaver, A.; Bren, A.; Ronen, M.; Itzkovitz, S.; Kikoin, I.; Shavit, S.; Liebermeister, W.; Surette, M.G.; Alon, U. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat. Methods 2006, 3, 623–628. [Google Scholar] [CrossRef]
- Amato, S.M.; Brynildsen, M.P. Nutrient Transitions Are a Source of Persisters in Escherichia coli Biofilms. PLoS ONE 2014, 9, e93110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, S.M.; Brynildsen, M.P. Persister Heterogeneity Arising from a Single Metabolic Stress. Curr. Biol. 2015, 25, 2090–2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640. [Google Scholar] [CrossRef] [Green Version]
- Cherepanov, P.P.; Wackernagel, W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 1995, 158, 9–14. [Google Scholar] [CrossRef]
- Miller, J.H. Experiments in Molecular Genetics; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1972; p. 466. [Google Scholar]
- Griffith, K.L.; Wolf, R.E. Measuring β-Galactosidase Activity in Bacteria: Cell Growth, Permeabilization, and Enzyme Assays in 96-Well Arrays. Biochem. Biophys. Res. Commun. 2002, 290, 397–402. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Chou, W.K.; Brynildsen, M.P. Loss of DksA leads to multi-faceted impairment of nitric oxide detoxification by Escherichia coli. Free Radic. Biol. Med. 2019, 130, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Reis, A.C.; Salis, H.M. An Automated Model Test System for Systematic Development and Improvement of Gene Expression Models. ACS Synth. Biol. 2020, 9, 3145–3156. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.K.; Vaikunthan, M.; Schröder, H.V.; Link, A.J.; Kim, H.; Brynildsen, M.P. Synergy Screening Identifies a Compound That Selectively Enhances the Antibacterial Activity of Nitric Oxide. Front. Bioeng. Biotechnol. 2020, 8, 1001. [Google Scholar] [CrossRef]
- Głąb, T.K.; Boratyński, J. Potential of Casein as a Carrier for Biologically Active Agents. Top. Curr. Chem. 2017, 375, 71. [Google Scholar] [CrossRef] [Green Version]
- Fahnert, B.; Lilie, H.; Neubauer, P. Inclusion Bodies: Formation and Utilisation. In Physiological Stress Responses in Bioprocesses; Springer: Berlin/Heidelberg, Germany, 2004; pp. 93–142. [Google Scholar]
- Villaverde, A.; Mar Carrió, M. Protein aggregation in recombinant bacteria: Biological role of inclusion bodies. Biotechnol. Lett. 2003, 25, 1385–1395. [Google Scholar] [CrossRef] [PubMed]
- Salis, H.M. Chapter two—The Ribosome Binding Site Calculator. In Methods in Enzymology; Voigt, C., Ed.; Academic Press: Cambridge, MA, USA, 2011; Volume 498, pp. 19–42. [Google Scholar]
- The Business Research Company. Therapeutic Proteins Global Market Report 2021: COVID-19 Impact and Recovery to 2030; The Business Research Company: London, UK, 2021. [Google Scholar]
- Belsey, M.; Pavlou, A. Marketspace: Leading therapeutic recombinant protein sales forecast and analysis to 2010. J. Commer. Biotechnol. 2005, 12, 69–73. [Google Scholar] [CrossRef]
- Dewan, S.S. Global Markets for Enzymes in Industrial Applications; BCC Publishing: Wellesley, MA, USA, 2021; p. 169. [Google Scholar]
- Young, C.L.; Britton, Z.T.; Robinson, A.S. Recombinant protein expression and purification: A comprehensive review of affinity tags and microbial applications. Biotechnol. J. 2012, 7, 620–634. [Google Scholar] [CrossRef] [PubMed]
- di Guana, C.; Lib, P.; Riggsa, P.D.; Inouyeb, H. Vectors that facilitate the expression and purification of foreign peptides in Escherichia coli by fusion to maltose-binding protein. Gene 1988, 67, 21–30. [Google Scholar] [CrossRef]
- Kapust, R.B.; Waugh, D.S. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 1999, 8, 1668–1674. [Google Scholar] [CrossRef] [Green Version]
- Davis, G.D.; Elisee, C.; Newham, D.M.; Harrison, R.G. New fusion protein systems designed to give soluble expression in Escherichia coli. Biotechnol. Bioeng. 1999, 65, 382–388. [Google Scholar] [CrossRef]
- LaVallie, E.R.; Lu, Z.; Diblasio-Smith, E.A.; Collins-Racie, L.A.; McCoy, J.M. [21] Thioredoxin as a fusion partner for production of soluble recombinant proteins in Escherichia coli. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2000; Volume 326, pp. 322–340. [Google Scholar]
- Chaves, J.E.; Rueda-Romero, P.; Kirst, H.; Melis, A. Engineering Isoprene Synthase Expression and Activity in Cyanobacteria. ACS Synth. Biol. 2017, 6, 2281–2292. [Google Scholar] [CrossRef]
- Formighieri, C.; Melis, A. A phycocyanin·phellandrene synthase fusion enhances recombinant protein expression and β-phellandrene (monoterpene) hydrocarbons production in Synechocystis (cyanobacteria). Metab. Eng. 2015, 32, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Butt, T.R.; Edavettal, S.C.; Hall, J.P.; Mattern, M.R. SUMO fusion technology for difficult-to-express proteins. Protein Expr. Purif. 2005, 43, 1–9. [Google Scholar] [CrossRef]
- Smith, D.B.; Johnson, K.S. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 1988, 67, 31–40. [Google Scholar] [CrossRef]
- Waugh, D.S. Making the most of affinity tags. Trends Biotechnol. 2005, 23, 316–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in microbial systems. Front. Microbiol. 2014, 5, 341. [Google Scholar] [CrossRef] [Green Version]
- Ilari, A.; Bonamore, A.; Farina, A.; Johnson, K.A.; Boffi, A. The X-ray Structure of Ferric Escherichia coli Flavohemoglobin Reveals an Unexpected Geometry of the Distal Heme Pocket. J. Biol. Chem. 2002, 277, 23725–23732. [Google Scholar] [CrossRef] [Green Version]
- Ahn, K.-Y.; Song, J.-A.; Han, K.-Y.; Park, J.-S.; Seo, H.-S.; Lee, J. Heterologous protein expression using a novel stress-responsive protein of E. coli RpoA as fusion expression partner. Enzym. Microb. Technol. 2007, 41, 859–866. [Google Scholar] [CrossRef]
- Han, K.-Y.; Song, J.-A.; Ahn, K.-Y.; Park, J.-S.; Seo, H.-S.; Lee, J. Solubilization of aggregation-prone heterologous proteins by covalent fusion of stress-responsive Escherichia coli protein, SlyD. Protein Eng. Des. Sel. 2007, 20, 543–549. [Google Scholar] [CrossRef] [Green Version]
- Han, K.-Y.; Song, J.-A.; Ahn, K.-Y.; Park, J.-S.; Seo, H.-S.; Lee, J. Enhanced solubility of heterologous proteins by fusion expression using stress-induced Escherichia coli protein, Tsf. FEMS Microbiol. Lett. 2007, 274, 132–138. [Google Scholar] [CrossRef]
- Park, J.-S.; Han, K.-Y.; Lee, J.-H.; Song, J.-A.; Ahn, K.-Y.; Seo, H.-S.; Sim, S.-J.; Kim, S.-W.; Lee, J. Solubility enhancement of aggregation-prone heterologous proteins by fusion expression using stress-responsive Escherichia coli protein, RpoS. BMC Biotechnol. 2008, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Han, K.-Y.; Seo, H.-S.; Song, J.-A.; Ahn, K.-Y.; Park, J.-S.; Lee, J. Transport proteins PotD and Crr of Escherichia coli, novel fusion partners for heterologous protein expression. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2007, 1774, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Pédelacq, J.-D.; Cabantous, S.; Tran, T.; Terwilliger, T.C.; Waldo, G.S. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 2006, 24, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Simons, G.; van den Heuvel, W.; Reynen, T.; Frijters, A.; Rutten, G.; Slangen, C.J.; Groenen, M.; de Vos, W.M.; Siezen, R.J. Overproduction of bovine β-casein in Escherichia coli and engineering of its main chymosin cleavage site. Protein Eng. Des. Sel. 1993, 6, 763–770. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, X.; Link, A.J.; Brynildsen, M.P. Translational Fusion to Hmp Improves Heterologous Protein Expression. Microorganisms 2022, 10, 358. https://doi.org/10.3390/microorganisms10020358
Wan X, Link AJ, Brynildsen MP. Translational Fusion to Hmp Improves Heterologous Protein Expression. Microorganisms. 2022; 10(2):358. https://doi.org/10.3390/microorganisms10020358
Chicago/Turabian StyleWan, Xuanqing, A. James Link, and Mark P. Brynildsen. 2022. "Translational Fusion to Hmp Improves Heterologous Protein Expression" Microorganisms 10, no. 2: 358. https://doi.org/10.3390/microorganisms10020358
APA StyleWan, X., Link, A. J., & Brynildsen, M. P. (2022). Translational Fusion to Hmp Improves Heterologous Protein Expression. Microorganisms, 10(2), 358. https://doi.org/10.3390/microorganisms10020358