
Phage Genome Diversity in a Biogas-Producing Microbiome Analyzed by Illumina and Nanopore GridION Sequencing

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Reactor Set-Up and Sampling
2.2. Electron Microscopy
2.3. Enrichment and Purification of Phage Particles from a Biogas Microbiome
2.4. Phage DNA Extraction
2.5. MinION Library Preparation and Sequencing
2.6. Illumina Library Preparation and Sequencing
2.7. Base Calling, Read Processing, and Assembly
2.8. Virome Filtering, Annotation, and Genome Analysis
2.9. Fragment Recruitment
3. Results and Discussion
3.1. Occurrence of Virus-Like Particles in the Studied Biogas Reactor by Means of Transmission Electron Microscopy
3.2. Virome Separation, Enrichment, Sequencing, and Bioinformatic Analysis
3.3. Phage Diversity in the Studied Biogas Plant as Analyzed by Means of Viral Metagenome Data
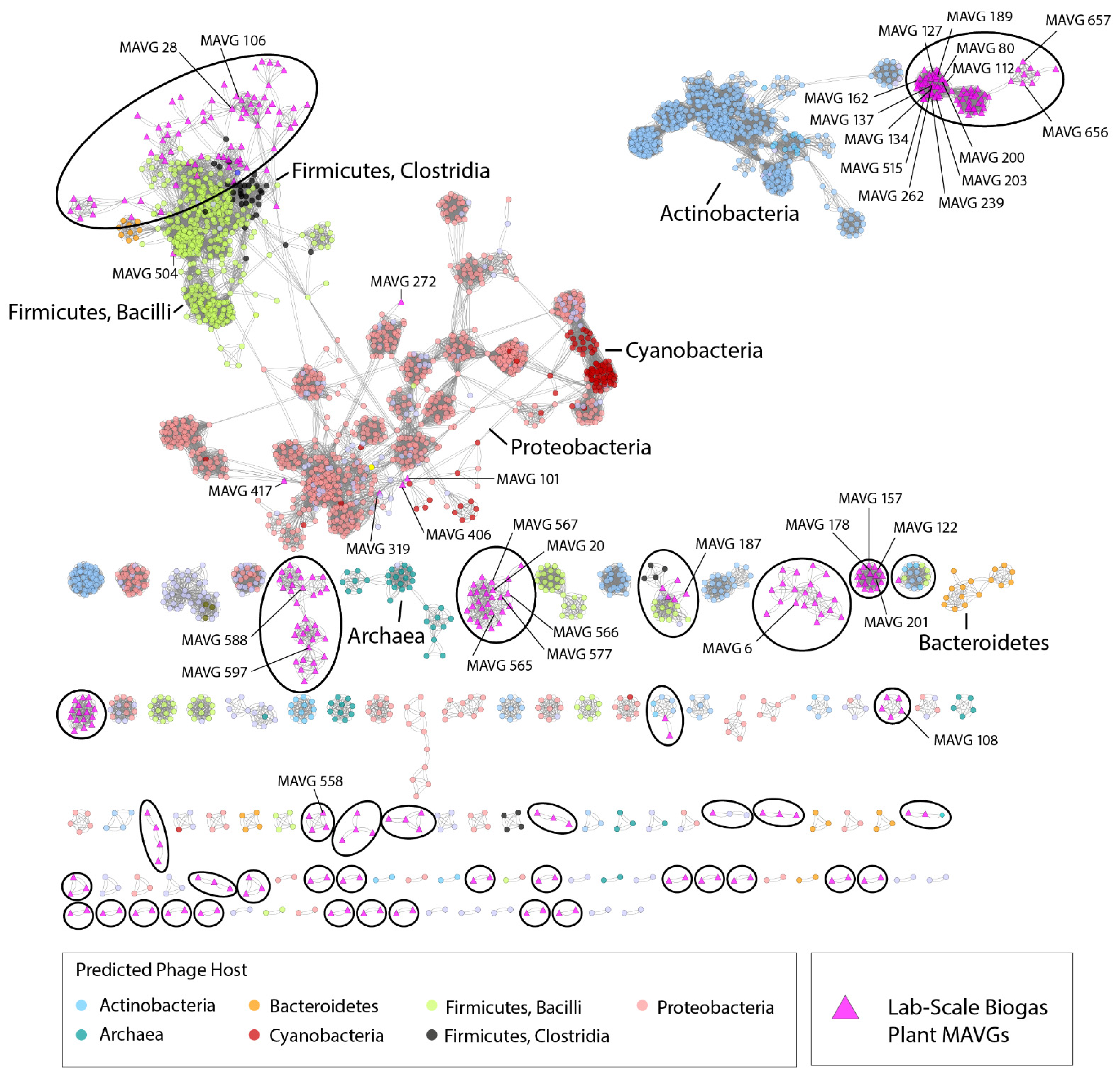
3.4. Protein-Based Phage Similarity Network Showed Relationships with Phages Infecting Members of the Classes Bacilli and Clostridia
3.5. Insights into the Biogas Plant Phages Life Cycle by Comprehensive Genome Analysis
3.6. Occurrence of Phage Relatives in Biogas-Producing Microbial Communities as Deduced from Publicly Available Metagenome Data
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suttle, C. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Prangishvili, D.; Hendrix, R.W.; Bamford, D.H. Genomics of Bacterial and Archaeal Viruses: Dynamics within the Prokaryotic Virosphere. Microbiol. Mol. Biol. Rev. 2011, 75, 610–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suttle, C. Marine viruses--major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Bonnain, C.; Malki, K. Sawaya Phage puppet masters of the marine microbial realm. Nat. Microbiol. 2018, 3, 754–766. [Google Scholar] [CrossRef]
- Weitz, J.S.; Stock, C.A.; Wilhelm, S.W.; Bourouiba, L.; Coleman, M.L.; Buchan, A.; Follows, M.J.; Fuhrman, J.A.; Jover, L.F.; Lennon, J.T.; et al. A multitrophic model to quantify the effects of marine viruses on microbial food webs and ecosystem processes. ISME J. 2015, 9, 1352–1364. [Google Scholar] [CrossRef] [Green Version]
- Bragg, J.G.; Chisholm, S.W. Modeling the Fitness Consequences of a Cyanophage-Encoded Photosynthesis Gene. PLoS ONE 2008, 3, e3550. [Google Scholar] [CrossRef] [Green Version]
- Mann, N.H.; Cook, A.; Millard, A.; Bailey, S.; Clokie, M. Bacterial photosynthesis genes in a virus. Nature 2003, 424, 741. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.X.; Méheust, R.; Crits-Christoph, A.; McMahon, K.D.; Nelson, T.C.; Slater, G.F.; Warren, L.A.; Banfield, J.F. Large freshwater phages with the potential to augment aerobic methane oxidation. Nat. Microbiol. 2020, 5, 1504–1515. [Google Scholar] [CrossRef]
- Ahlgren, N.A.; Fuchsman, C.A.; Rocap, G.; Fuhrman, J.A. Discovery of several novel, widespread, and ecologically distinct marine Thaumarchaeota viruses that encode amoC nitrification genes. ISME J. 2018, 13, 618–631. [Google Scholar] [CrossRef] [Green Version]
- Hernández, S.; Vives, M. Phages in Anaerobic Systems. Viruses 2020, 12, 1091. [Google Scholar] [CrossRef]
- Strange, J.E.; Leekitcharoenphon, P.; Duus Møller, F.; Aarestrup, F.M. Metagenomics analysis of bacteriophages and antimicrobial resistance from global urban sewage. Sci. Rep. 2021, 11, 1600. [Google Scholar] [CrossRef]
- Camarillo-Guerrero, L.F.; Almeida, A.; Rangel-Pineros, G.; Finn, R.D.; Lawley, T.D. Massive expansion of human gut bacteriophage diversity. Cell 2021, 184, 1098–1109.e9. [Google Scholar] [CrossRef]
- Mcshan, W.M.; Lafont, B.A.P.; Bertelli, C.; Van Niftrik, L.; Gambelli, L.; Cremers, G.; Mesman, R.; Guerrero, S.; Dutilh, B.E.; Jetten, M.S.M.; et al. Ultrastructure and Viral Metagenome of Bacteriophages from an Anaerobic Methane Oxidizing Methylomirabilis Bioreactor Enrichment Culture. Front Microbiol. Front Org. 2016, 1, 1740. [Google Scholar] [CrossRef] [Green Version]
- Braga, L.P.P.; Spor, A.; Kot, W.; Breuil, M.-C.; Hansen, L.H.; Setubal, J.C.; Philippot, L. Impact of phages on soil bacterial communities and nitrogen availability under different assembly scenarios. Microbiome 2020, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tikhe, C.V.; Husseneder, C. Metavirome sequencing of the termite gut reveals the presence of an unexplored bacteriophage community. Front Microbiol. 2018, 8, 2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewandowski, I.; Bahrs, E.; Dahmen, N.; Hirth, T.; Rausch, T.; Weidtmann, A. Biobased value chains for a growing bioeconomy. GCB Bioenergy 2019, 11, 4–8. [Google Scholar] [CrossRef]
- Heyer, R.; Schallert, K.; Siewert, C.; Kohrs, F.; Greve, J.; Maus, I.; Klang, J.; Klocke, M.; Heiermann, M.; Hoffmann, M.; et al. Metaproteome analysis reveals that syntrophy, competition, and phage-host interaction shape microbial communities in biogas plants. Microbiome 2019, 7, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Solli, L.; Håvelsrud, O.E.; Horn, S.J.; Rike, A.G. A metagenomic study of the microbial communities in four parallel biogas reactors. Biotechnol. Biofuels 2014, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Maus, I.; Bremges, A.; Stolze, Y.; Hahnke, S.; Cibis, K.G.; Koeck, D.E.; Kim, Y.S.; Kreubel, J.; Hassa, J.; Wibberg, D.; et al. Genomics and prevalence of bacterial and archaeal isolates from biogas-producing microbiomes. Biotechnol. Biofuels 2017, 10, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Stolze, Y.; Bremges, A.; Maus, I.; Pühler, A.; Sczyrba, A.; Schlüter, A. Targeted in situ metatranscriptomics for selected taxa from mesophilic and thermophilic biogas plants. Microb. Biotechnol. 2018, 11, 667–679. [Google Scholar] [CrossRef] [Green Version]
- Campanaro, S.; Treu, L.; Rodriguez-R, L.M.; Kovalovszki, A.; Ziels, R.M.; Maus, I.; Zhu, X.; Kougias, P.G.; Basile, A.; Luo, G.; et al. New insights from the biogas microbiome by comprehensive genome-resolved metagenomics of nearly 1600 species originating from multiple anaerobic digesters. Biotechnol. Biofuels 2020, 13, S13068–S020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, C.; Bongcam-Rudloff, E.; Müller, B. Abundance Tracking by Long-Read Nanopore Sequencing of Complex Microbial Communities in Samples from 20 Different Biogas/Wastewater Plants. Appl. Sci. 2020, 10, 7518. [Google Scholar] [CrossRef]
- Overholt, W.A.; Hölzer, M.; Geesink, P.; Diezel, C.; Marz, M.; Küsel, K. Inclusion of Oxford Nanopore long reads improves all microbial and viral metagenome-assembled genomes from a complex aquifer system. Environ. Microbiol. 2020, 22, 4000–4013. [Google Scholar] [CrossRef]
- Stolze, Y.; Bremges, A.; Rumming, M.; Henke, C.; Maus, I.; Pühler, A.; Sczyrba, A.; Schlüter, A. Identification and genome reconstruction of abundant distinct taxa in microbiomes from one thermophilic and three mesophilic production-scale biogas plants. Biotechnol. Biofuels 2016, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Maus, I.; Koeck, D.E.; Cibis, K.G.; Hahnke, S.; Kim, Y.S.; Langer, T.; Kreubel, J.; Erhard, M.; Bremges, A.; Off, S.; et al. Unraveling the microbiome of a thermophilic biogas plant by metagenome and metatranscriptome analysis complemented by characterization of bacterial and archaeal isolates. Biotechnol. Biofuels 2016, 9, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Maus, I.; Rumming, M.; Bergmann, I.; Heeg, K.; Pohl, M.; Nettmann, E.; Jaenicke, S.; Blom, J.; Pühler, A.; Schlüter, A.; et al. Characterization of Bathyarchaeota genomes assembled from metagenomes of biofilms residing in mesophilic and thermophilic biogas reactors. Biotechnol. Biofuels 2018, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Weiland, P. Biogas production: Current state and perspectives. Appl. Microbiol. Biotechnol. 2009, 85, 849–860. [Google Scholar] [CrossRef]
- Gaspari, M.; Treu, L.; Zhu, X.; Palù, M.; Angelidaki, I.; Campanaro, S.; Kougias, P. Microbial dynamics in biogas digesters treating lipid-rich substrates via genome-centric metagenomics. Sci. Total Environ. 2021, 778, 146296. [Google Scholar] [CrossRef]
- Calusinska, M.; Marynowska, M.; Goux, X.; Lentzen, E.; Delfosse, P. Analysis of dsDNA and RNA viromes in methanogenic digesters reveals novel viral genetic diversity. Environ. Microbiol. 2016, 18, 1162–1175. [Google Scholar] [CrossRef] [PubMed]
- Bujak, K.; Decewicz, P.; Kaminski, J.; Radlinska, M. Identification, Characterization, and Genomic Analysis of Novel Serratia Temperate Phages from a Gold Mine. Int. J. Mol. Sci. Artic. 2020, 21, 6709. [Google Scholar] [CrossRef]
- Pietilä, M.K.; Demina, T.A.; Atanasova, N.S.; Oksanen, H.M.; Bamford, D.H. Archaeal viruses and bacteriophages: Comparisons and contrasts. Trends Microbiol. 2014, 22, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Dion, M.; Oechslin, F.; Moineau, S. Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Chien, I.; Meschke, J.S.; Gough, H.L.; Ferguson, J.F. Characterization of persistent virus-like particles in two acetate-fed methanogenic reactors. PLoS ONE 2013, 8, e81040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamaki, H.; Zhang, R.; Angly, F.; Nakamura, S.; Hong, P.; Yasunaga, T.; Kamagata, Y.; Liu, W. Metagenomic analysis of DNA viruses in a wastewater treatment plant in tropical climate. Environ. Microbiol. 2012, 14, 441–452. [Google Scholar] [CrossRef]
- Shapiro, O.H.; Kushmaro, A.; Brenner, A. Bacteriophage predation regulates microbial abundance and diversity in a full-scale bioreactor treating industrial wastewater. ISME J. 2009, 4, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Gao, Q.; Zhang, Q.; Wang, T.; Yue, H.; Wu, L.; Shi, J.; Qin, Z.; Zhou, J.; Zuo, J.; et al. Bacteriophage–prokaryote dynamics and interaction within anaerobic digestion processes across time and space. Microbiome 2017, 5, 1–10. [Google Scholar] [CrossRef]
- Ackermann, H.W. Basic phage electron microscopy. Methods Mol. Biol. 2009, 501, 113–126. [Google Scholar] [CrossRef]
- Wibberg, D.; Stadler, M.; Lambert, C.; Bunk, B.; Spröer, C.; Rückert, C.; Kalinowski, J.; Cox, R.J.; Kuhnert, E. High quality genome sequences of thirteen Hypoxylaceae (Ascomycota) strengthen the phylogenetic family backbone and enable the discovery of new taxa. Fungal Divers. 2020, 106, 7–28. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Villarroel, J.; Kleinheinz, K.A.; Jurtz, V.I.; Zschach, H.; Lund, O.; Nielsen, M.; Larsen, M.V. HostPhinder: A Phage Host Prediction Tool. Viruses 2016, 8, 116. [Google Scholar] [CrossRef]
- Marquet, M.; Hölzer, M.; Pletz, M.W.; Viehweger, A.; Makarewicz, O.; Ehricht, R.; Brandt, C. What the Phage: A scalable workflow for the identification and analysis of phage sequences. bioRxiv 2020, 11, e00460-20. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Goesmann, A.; McHardy, A.C.; Bartels, D.; Bekel, T.; Clausen, J.; Kalinowski, J.; Linke, B.; Rupp, O.; Giegerich, R.; et al. GenDB—An open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 2003, 31, 2187–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; Volume 35, ISBN 978-0-387-98140-6. [Google Scholar]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, B.; Jang, H.B.; Doulcier, G.; You, Z.Q.; Roux, S.; Sullivan, M.B. vConTACT: An iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. Peer J. 2017, 5, e3243. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- McNair, K.; Bailey, B.A.; Edwards, R.A. PHACTS, a computational approach to classifying the lifestyle of phages. Bioinformatics 2012, 28, 614–618. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Krüger, J.; Sczyrba, A. Analyzing large scale genomic data on the cloud with Sparkhit. Bioinformatics 2018, 34, 1457–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maus, I.; Tubbesing, T.; Wibberg, D.; Heyer, R.; Hassa, J.; Tomazetto, G.; Huang, L.; Bunk, B.; Spröer, C.; Benndorf, D.; et al. The Role of Petrimonas mucosa ING2-E5AT in Mesophilic Biogas Reactor Systems as Deduced from Multiomics Analyses. Microorgism 2020, 8, 2024. [Google Scholar] [CrossRef] [PubMed]
- Verein Deutscher Ingenieure (VDI) 4630: Fermentation of Organic Materials—Characterisation of the Substrate, Sampling, Collection of Material Data, Fermentation Tests; Beuth Verlag: Berlin, Germany, 2016; p. 132.
- Keating, C.; Cysneiros, D.; Mahony, T.; O’Flaherty, V. The hydrolysis and biogas production of complex cellulosic substrates using three anaerobic biomass sources. Water Sci. Technol. 2013, 67, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Decewicz, P.; Golec, P.; Szymczak, M.; Radlinska, M.; Dziewit, L. Identification and characterization of the first virulent phages, including a novel jumbo virus, infecting Ochrobactrum spp. Int. J. Mol. Sci. 2020, 21, 2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyer, R.; Schallert, K.; Zoun, R.; Becher, B.; Saake, G.; Benndorf, D. Challenges and perspectives of metaproteomic data analysis. J. Biotechnol. 2017, 261, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Weidenbach, K.; Wolf, S.; Kupczok, A.; Kern, T.; Fischer, M.A.; Reetz, J.; Urbańska, N.; Künzel, S.; Schmitz, R.A.; Rother, M. Characterization of Blf4, an Archaeal Lytic Virus Targeting a Member of the Methanomicrobiales. Viruses 2021, 13, 1934. [Google Scholar] [CrossRef] [PubMed]
- Stolze, Y.; Zakrzewski, M.; Maus, I.; Eikmeyer, F.; Jaenicke, S.; Rottmann, N.; Siebner, C.; Pühler, A.; Schlüter, A. Comparative metagenomics of biogas-producing microbial communities from production-scale biogas plants operating under wet or dry fermentation conditions. Biotechnol. Biofuels 2015, 8, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maus, I.; Wibberg, D.; Stantscheff, R.; Stolze, Y.; Blom, J.; Eikmeyer, F.G.; Fracowiak, J.; König, H.; Pühler, A.; Schlüter, A. Insights into the annotated genome sequence of Methanoculleus bourgensis MS2(T), related to dominant methanogens in biogas-producing plants. J. Biotechnol. 2015, 201, 43–53. [Google Scholar] [CrossRef]
- Fornelos, N.; Browning, D.F.; Pavlin, A.; Podlesek, Z.; Hodnik, V.; Salas, M.; Butala, M. Lytic gene expression in the temperate bacteriophage GIL01 is activated by a phage-encoded LexA homologue. Nucleic Acids Res. 2018, 46, 9432–9443. [Google Scholar] [CrossRef]
- Krupovic, M.; Cvirkaite-Krupovic, V.; Prangishvili, D.; Koonin, E.V. Evolution of an archaeal virus nucleocapsid protein from the CRISPR-associated Cas4 nuclease. Biol. Direct 2015, 10, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, R.E. Filamentous phages (inoviridae). Encycl. Virol. 1999, 2, 547–552. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Sullivan, M.B.; Knezevic, P.; van Zyl, L.J.; Sarkar, B.L.; Dutilh, B.E.; Alfenas-Zerbini, P.; Łobocka, M.; Tong, Y.; Brister, J.R.; et al. Taxonomy of prokaryotic viruses: 2018-2019 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2020, 165, 1253–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, A.M.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. Virus Taxonomy. Ninth Rep. Int. Comm. Taxon. Viruses 2012, 9, 1–5. [Google Scholar]
- Tiemann, B.; Depping, R.; Rüger, W. Overexpression, Purification, and Partial Characterization of ADP-Ribosyltransferases ModA and ModB of Bacteriophage T4. Gene Expr. 1999, 8, 187. [Google Scholar]
- Brown, E.M.; Arellano-Santoyo, H.; Temple, E.R.; Costliow, Z.A.; Pichaud, M.; Hall, A.B.; Liu, K.; Durney, M.A.; Gu, X.; Plichta, D.R.; et al. Gut microbiome ADP-ribosyltransferases are widespread phage-encoded fitness factors. Cell Host Microbe 2021, 29, 1351–1365.e11. [Google Scholar] [CrossRef]
- Kumar, R.; Feltrup, T.M.; Kukreja, R.V.; Patel, K.B.; Cai, S.; Singh, B.R. Evolutionary Features in the Structure and Function of Bacterial Toxins. Toxins 2019, 11, 15. [Google Scholar] [CrossRef] [Green Version]
- Shmakov, S.A.; Makarova, K.S.; Wolf, Y.I.; Severinov, K.V.; Koonin, E.V. Systematic prediction of genes functionally linked to CRISPR-Cas systems by gene neighborhood analysis. Proc. Natl. Acad. Sci. USA 2018, 115, E5307–E5316. [Google Scholar] [CrossRef] [Green Version]
- Šulčius, S.; Alzbutas, G.; Juknevičiūtė, V.; Šimoliūnas, E.; Venckus, P.; Šimoliūnienė, M.; Paškauskas, R. Exploring Viral Diversity in a Gypsum Karst Lake Ecosystem Using Targeted Single-Cell Genomics. Genes 2021, 12, 886. [Google Scholar] [CrossRef]
- McKay, L.J.; Nigro, O.D.; Dlakić, M.; Luttrell, K.M.; Rusch, D.B.; Fields, M.W.; Inskeep, W.P. Sulfur cycling and host-virus interactions in Aquificales-dominated biofilms from Yellowstone’s hottest ecosystems. ISME J. 2021, 1–14. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 80 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Güllert, S.; Fischer, M.A.; Turaev, D.; Noebauer, B.; Ilmberger, N.; Wemheuer, B.; Alawi, M.; Rattei, T.; Daniel, R.; Schmitz, R.A.; et al. Deep metagenome and metatranscriptome analyses of microbial communities affiliated with an industrial biogas fermenter, a cow rumen, and elephant feces reveal major differences in carbohydrate hydrolysis strategies. Biotechnol. Biofuels 2016, 9, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position According to the MAVG Abundance | Phage Contig ID | Contig Length [bp] | Coverage | Circular Genome | Number of Mapped Sequences **** | Taxonomy at the Family Level |
|---|---|---|---|---|---|---|
| 1 | MAVG 80 | 12,554 | 9083 | no | 16,283 | Siphoviridae |
| 2 | MAVG 112 | 12,903 | 214 | no | 684 | Myoviridae |
| 3 | MAVG 134 | 11,568 | 162 | no | 498 | Siphoviridae |
| 4 | MAVG 567 | 10,561 | 156 | no | 428 | Myoviridae |
| 5 | MAVG 137 | 10,247 | 149 | no | 389 | Siphoviridae |
| 6 | MAVG 178 | 8780 | 90 | no | 214 | unclassified |
| 7 | MAVG 127 | 11,531 | 84 | no | 285 | Podoviridae |
| 8 | MAVG 108 | 12,998 | 79 | no | 271 | Siphoviridae |
| 9 | MAVG 189 | 10,460 | 63 | no | 186 | unclassified |
| 10 | MAVG 566 | 10,816 | 54 | no | 142 | unclassified |
| 11 | MAVG 657 | 7709 | 52 | no | 136 | unclassified |
| 12 | MAVG 200 | 7316 | 47 | no | 99 | Myoviridae |
| 13 | MAVG 28 | 44,297 | 45 | yes | 560 | Myoviridae |
| 14 | MAVG 187 | 8926 | 43 | no | 133 | Siphoviridae |
| 15 | MAVG 577 | 9334 | 42 | no | 98 | unclassified |
| 16 | MAVG 203 | 8666 | 42 | no | 98 | Siphoviridae |
| 17 | MAVG 122 | 9252 | 42 | no | 112 | Siphoviridae |
| 18 | MAVG 20 | 10,140 | 40 | no | 121 | unclassified |
| 19 | MAVG 239 | 9882 | 40 | no | 117 | unclassified |
| 20 | MAVG 21 | 8028 | 39 | yes | 84 | Siphoviridae |
| 21 | MAVG 201 | 9048 | 37 | no | 104 | Siphoviridae |
| 22 | MAVG 597 | 31,553 | 37 | no | 323 | unclassified |
| 23 | MAVG 162 | 5791 | 35 | no | 96 | Siphoviridae |
| 24 | MAVG 222 | 8975 | 34 | no | 90 | Myoviridae |
| 25 | MAVG 593 | 36,077 | 34 | no | 405 | Myoviridae |
| 26 | MAVG 202 | 8422 | 34 | no | 77 | Myoviridae |
| 27 | MAVG 252 | 9143 | 34 | no | 94 | Siphoviridae |
| 28 | MAVG 160 | 10,174 | 33 | no | 96 | Siphoviridae |
| 98 | MAVG 6 * | 63,815 | 13 | yes | 250 | Siphoviridae |
| 37 | MAVG 588 * | 52,688 | 29 | yes | 1 | unclassified |
| 51 | MAVG 157 ** | 8517 | 24 | yes | 4 | Inoviridae |
| 94 | MAVG 515 *** | 5271 | 14 | yes | 2 | Tectiviridae |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willenbücher, K.; Wibberg, D.; Huang, L.; Conrady, M.; Ramm, P.; Gätcke, J.; Busche, T.; Brandt, C.; Szewzyk, U.; Schlüter, A.; et al. Phage Genome Diversity in a Biogas-Producing Microbiome Analyzed by Illumina and Nanopore GridION Sequencing. Microorganisms 2022, 10, 368. https://doi.org/10.3390/microorganisms10020368
Willenbücher K, Wibberg D, Huang L, Conrady M, Ramm P, Gätcke J, Busche T, Brandt C, Szewzyk U, Schlüter A, et al. Phage Genome Diversity in a Biogas-Producing Microbiome Analyzed by Illumina and Nanopore GridION Sequencing. Microorganisms. 2022; 10(2):368. https://doi.org/10.3390/microorganisms10020368
Chicago/Turabian StyleWillenbücher, Katharina, Daniel Wibberg, Liren Huang, Marius Conrady, Patrice Ramm, Julia Gätcke, Tobias Busche, Christian Brandt, Ulrich Szewzyk, Andreas Schlüter, and et al. 2022. "Phage Genome Diversity in a Biogas-Producing Microbiome Analyzed by Illumina and Nanopore GridION Sequencing" Microorganisms 10, no. 2: 368. https://doi.org/10.3390/microorganisms10020368