
DNA Enrichment Methods for Microbial Symbionts in Marine Bivalves

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Treatment
2.2. DNA Extraction
2.3. Metagenomic Sequencing and Data Analysis
2.4. 16S rRNA Gene Amplicon Sequencing and Analysis
3. Results
3.1. Quality and Quantity of the Extracted DNA
3.2. Efficiency of Host DNA Depletion
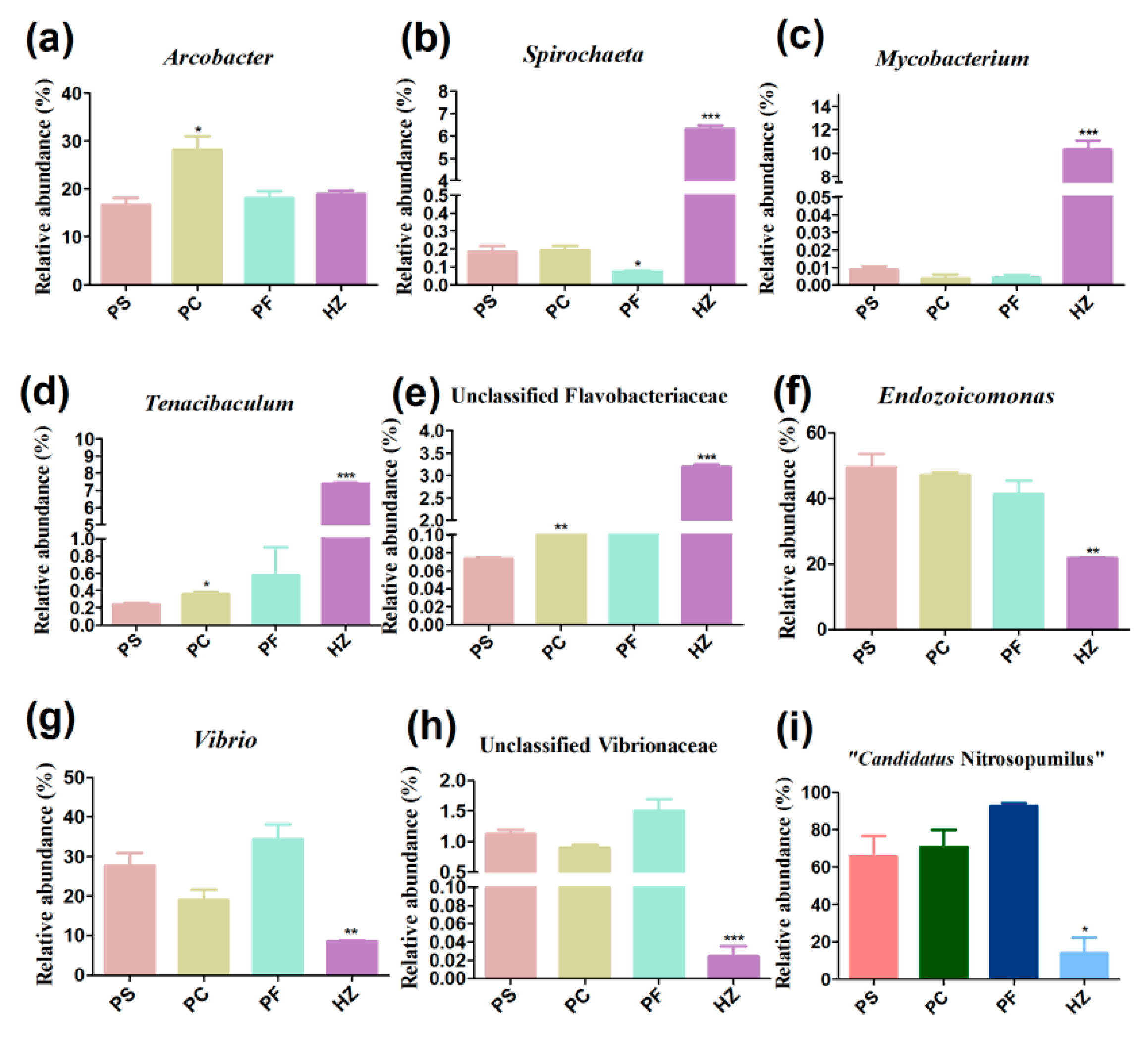
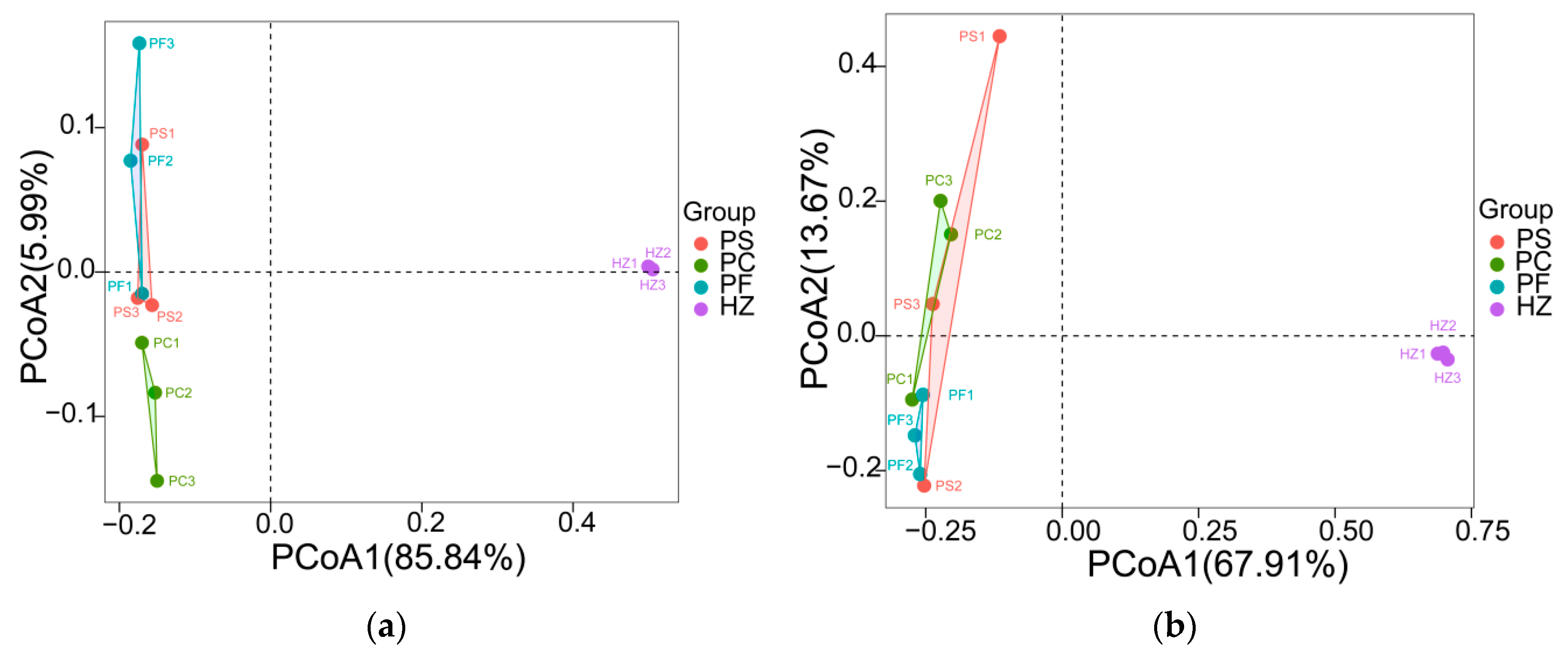
3.3. Compositions and Diversities of Bacterial Communities Retrieved Using Different Methods
3.4. Compositions and Diversities of Archaeal Communities Retrieved Using Different Methods
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Distel, D.L.; Baco, A.R.; Chuang, E.; Morrill, W.; Cavanaugh, C.; Smith, C.R. Do mussels take wooden steps to deep-sea vents? Nature 2000, 403, 725–726. [Google Scholar] [CrossRef]
- Bogan, A.E. Global diversity of freshwater mussels (Mollusca, Bivalvia) in freshwater. Hydrobiologia 2008, 595, 139–147. [Google Scholar] [CrossRef]
- Duperron, S.; Halary, S.; Lorion, J.; Sibuet, M.; Gaill, F.J.E.M. Unexpected co-occurrence of six bacterial symbionts in the gills of the cold seep mussel Idas sp. (Bivalvia: Mytilidae). Environ. Microbiol. 2008, 10, 433–445. [Google Scholar] [CrossRef]
- Distel, D.L.; Altamia, M.A.; Lin, Z.; Shipway, J.R.; Han, A.; Forteza, I.; Antemano, R.; Limbaco, M.; Tebo, A.G.; Dechavez, R.; et al. Discovery of chemoautotrophic symbiosis in the giant shipworm Kuphus polythalamia (Bivalvia: Teredinidae) extends wooden-steps theory. Proc. Natl. Acad. Sci. USA 2017, 114, E3652–E3658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duperron, S.; Gaudron, S.; Rodrigues, C.; Cunha, M.; Decker, C.; Olu, K.J.B. An overview of chemosynthetic symbioses in bivalves from the North Atlantic and Mediterranean Sea. Biogeosci. Discuss. 2013, 10, 3241–3267. [Google Scholar] [CrossRef] [Green Version]
- Rhoads, D.C.; Lutz, R.A.; Revelas, E.C.; Cerrato, R.M.J.S. Growth of bivalves at deep-sea hydrothermal vents along the Galapagos Rift. Science 1981, 214, 911–913. [Google Scholar] [CrossRef]
- Childress, J.J.; Fisher, C.; Brooks, J.; Kennicutt, M.; Bidigare, R.; Anderson, A.J.S. A methanotrophic marine molluscan (Bivalvia, Mytilidae) symbiosis: Mussels fueled by gas. Science 1986, 233, 1306–1308. [Google Scholar] [CrossRef]
- Smith, C.R.; Kukert, H.; Wheatcroft, R.A.; Jumars, P.A.; Deming, J.W.J.N. Vent fauna on whale remains. Nature 1989, 341, 27–28. [Google Scholar] [CrossRef]
- Dell, R.K. Mollusca of the family Mytilidae (Bivalvia) associated with organic remains from deep water off New Zealand, with revisions of the genera Adipicola Dautzenberg, 1927 and Idasola Iredale, 1915. Natl. Mus. NZ Rec. 1987, 3, 17–36. [Google Scholar]
- Schweimanns, M.; Felbeck, H. Significance of the occurrence of chemoautotrophic bacterial endosymbionts in lucinid clams from Bermuda. Mar. Ecol. Prog. Ser. 1985, 24, 113–120. [Google Scholar] [CrossRef]
- van der Heide, T.; Govers, L.L.; de Fouw, J.; Olff, H.; van der Geest, M.; van Katwijk, M.M.; Piersma, T.; van de Koppel, J.; Silliman, B.R.; Smolders, A.J.; et al. A three-stage symbiosis forms the foundation of seagrass ecosystems. Science 2012, 336, 1432–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubilier, N.; Bergin, C.; Lott, C. Symbiotic diversity in marine animals: The art of harnessing chemosynthesis. Nat. Rev. Microbiol. 2008, 6, 725–740. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.; Romao, C.V.; Goulart, J.; Cerqueira, T.; Santos, R.S.; Bettencourt, R. Activity of antioxidant enzymes in response to atmospheric pressure induced physiological stress in deep-sea hydrothermal vent mussel Bathymodiolus azoricus. Mar. Environ. Res. 2016, 114, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Company, R.; Serafim, A.; Cosson, R.P.; Fiala-Medioni, A.; Camus, L.; Serrao-Santos, R.; Joao Bebianno, M. Sub-lethal effects of cadmium on the antioxidant defence system of the hydrothermal vent mussel Bathymodiolus azoricus. Ecotoxicol. Environ. Saf. 2010, 73, 788–795. [Google Scholar] [CrossRef]
- Martins, E.; Figueras, A.; Novoa, B.; Santos, R.S.; Moreira, R.; Bettencourt, R. Comparative study of immune responses in the deep-sea hydrothermal vent mussel Bathymodiolus azoricus and the shallow-water mussel Mytilus galloprovincialis challenged with Vibrio bacteria. Fish Shellfish Immunol. 2014, 40, 485–499. [Google Scholar] [CrossRef]
- Bettencourt, R.; Roch, P.; Stefanni, S.; Rosa, D.; Colaco, A.; Santos, R.S. Deep sea immunity: Unveiling immune constituents from the hydrothermal vent mussel Bathymodiolus azoricus. Mar. Environ. Res. 2007, 64, 108–127. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Zhang, Y.; Xu, T.; Zhang, Y.; Mu, H.; Zhang, Y.; Lan, Y.; Fields, C.J.; Hui, J.H.L.; Zhang, W.; et al. Adaptation to deep-sea chemosynthetic environments as revealed by mussel genomes. Nat. Ecol. Evol. 2017, 1, 121. [Google Scholar] [CrossRef] [Green Version]
- Ponnudurai, R.; Kleiner, M.; Sayavedra, L.; Petersen, J.M.; Moche, M.; Otto, A.; Becher, D.; Takeuchi, T.; Satoh, N.; Dubilier, N.; et al. Metabolic and physiological interdependencies in the Bathymodiolus azoricus symbiosis. ISME J. 2017, 11, 463–477. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhang, H.; Zhong, Z.; Sun, Y.; Wang, M.; Chen, H.; Zhou, L.; Cao, L.; Lian, C.; Li, C. Molecular analyses of the gill symbiosis of the bathymodiolin mussel Gigantidas platifrons. iScience 2021, 24, 101894. [Google Scholar] [CrossRef]
- Winkel, M.; Pjevac, P.; Kleiner, M.; Littmann, S.; Meyerdierks, A.; Amann, R.; Mussmann, M. Identification and activity of acetate-assimilating bacteria in diffuse fluids venting from two deep-sea hydrothermal systems. FEMS Microbiol. Ecol. 2014, 90, 731–746. [Google Scholar] [CrossRef] [Green Version]
- Geier, B.; Sogin, E.M.; Michellod, D.; Janda, M.; Kompauer, M.; Spengler, B.; Dubilier, N.; Liebeke, M. Spatial metabolomics of in situ host-microbe interactions at the micrometre scale. Nat. Microbiol. 2020, 5, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Franke, M.; Geier, B.; Hammel, J.U.; Dubilier, N.; Leisch, N. Coming together-symbiont acquisition and early development in deep-sea bathymodioline mussels. Proc. Royal Soc. B 2021, 288, 20211044. [Google Scholar] [CrossRef]
- Pathirana, E.; McPherson, A.; Whittington, R.; Hick, P. The role of tissue type, sampling and nucleic acid purification methodology on the inferred composition of Pacific oyster (Crassostrea gigas) microbiome. J. Appl. Microbiol. 2019, 127, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Heravi, F.S.; Zakrzewski, M.; Vickery, K.; Hu, H. Host DNA depletion efficiency of microbiome DNA enrichment methods in infected tissue samples. J. Microbiol. Methods 2020, 170, 105856. [Google Scholar] [CrossRef] [PubMed]
- Zaheer, R.; Noyes, N.; Ortega Polo, R.; Cook, S.R.; Marinier, E.; Van Domselaar, G.; Belk, K.E.; Morley, P.S.; McAllister, T.A. Impact of sequencing depth on the characterization of the microbiome and resistome. Sci. Rep. 2018, 8, 5890. [Google Scholar] [CrossRef]
- Ross, M.G.; Russ, C.; Costello, M.; Hollinger, A.; Lennon, N.J.; Hegarty, R.; Nusbaum, C.; Jaffe, D.B. Characterizing and measuring bias in sequence data. Genome Biol. 2013, 14, R51. [Google Scholar] [CrossRef] [Green Version]
- Snyder, M.; Du, J.; Gerstein, M. Personal genome sequencing: Current approaches and challenges. Genes Dev. 2010, 24, 423–431. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Su, X.; Jing, G.; Ning, K. Meta-QC-Chain: Comprehensive and fast quality control method for metagenomic data. Genom. Proteom. Bioinform. 2014, 12, 52–56. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef] [Green Version]
- Assie, A.; Leisch, N.; Meier, D.V.; Gruber-Vodicka, H.; Tegetmeyer, H.E.; Meyerdierks, A.; Kleiner, M.; Hinzke, T.; Joye, S.; Saxton, M.; et al. Horizontal acquisition of a patchwork Calvin cycle by symbiotic and free-living Campylobacterota (formerly Epsilonproteobacteria). ISME J. 2020, 14, 104–122. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.F.; Xu, J.K.; Chen, Y.W.; Ding, W.Y.; Shao, A.Q.; Liang, X.; Zhu, Y.T.; Yang, J.L. Characterization of Gut Microbiome in the Mussel Mytilus galloprovincialis in Response to Thermal Stress. Front. Physiol. 2019, 10, 1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weingarten, E.A.; Atkinson, C.L.; Jackson, C.R. The gut microbiome of freshwater Unionidae mussels is determined by host species and is selectively retained from filtered seston. PLoS ONE 2019, 14, e0224796. [Google Scholar] [CrossRef] [PubMed]
- Feehery, G.R.; Yigit, E.; Oyola, S.O.; Langhorst, B.W.; Schmidt, V.T.; Stewart, F.J.; Dimalanta, E.T.; Amaral-Zettler, L.A.; Davis, T.; Quail, M.A.; et al. A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PLoS ONE 2013, 8, e76096. [Google Scholar] [CrossRef] [PubMed]
- Kostic, T.; Weilharter, A.; Rubino, S.; Delogu, G.; Uzzau, S.; Rudi, K.; Sessitsch, A.; Bodrossy, L. A microbial diagnostic microarray technique for the sensitive detection and identification of pathogenic bacteria in a background of nonpathogens. Anal. Biochem. 2007, 360, 244–254. [Google Scholar] [CrossRef]
- Horz, H.P.; Scheer, S.; Vianna, M.E.; Conrads, G. New methods for selective isolation of bacterial DNA from human clinical specimens. Anaerobe 2010, 16, 47–53. [Google Scholar] [CrossRef]
- Ponnudurai, R.; Heiden, S.E.; Sayavedra, L.; Hinzke, T.; Kleiner, M.; Hentschker, C.; Felbeck, H.; Sievert, S.M.; Schluter, R.; Becher, D.; et al. Comparative proteomics of related symbiotic mussel species reveals high variability of host-symbiont interactions. ISME J. 2020, 14, 649–656. [Google Scholar] [CrossRef]
- Hinzke, T.; Kleiner, M.; Markert, S. Centrifugation-Based Enrichment of Bacterial Cell Populations for Metaproteomic Studies on Bacteria-Invertebrate Symbioses. Methods Mol. Biol. 2018, 1841, 319–334. [Google Scholar] [CrossRef]
- Ansorge, R.; Romano, S.; Sayavedra, L.; Porras, M.A.G.; Kupczok, A.; Tegetmeyer, H.E.; Dubilier, N.; Petersen, J. Functional diversity enables multiple symbiont strains to coexist in deep-sea mussels. Nat. Microbiol. 2019, 4, 2487–2497. [Google Scholar] [CrossRef]
- Kuwahara, H.; Yoshida, T.; Takaki, Y.; Shimamura, S.; Nishi, S.; Harada, M.; Matsuyama, K.; Takishita, K.; Kawato, M.; Uematsu, K.; et al. Reduced genome of the thioautotrophic intracellular symbiont in a deep-sea clam, Calyptogena okutanii. Curr. Biol. 2007, 17, 881–886. [Google Scholar] [CrossRef] [Green Version]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [Green Version]
- Ahannach, S.; Delanghe, L.; Spacova, I.; Wittouck, S.; Van Beeck, W.; De Boeck, I.; Lebeer, S. Microbial enrichment and storage for metagenomics of vaginal, skin, and saliva samples. iScience 2021, 24, 103306. [Google Scholar] [CrossRef] [PubMed]
- Grutzke, J.; Gwida, M.; Deneke, C.; Brendebach, H.; Projahn, M.; Schattschneider, A.; Hofreuter, D.; El-Ashker, M.; Malorny, B.; Al Dahouk, S. Direct identification and molecular characterization of zoonotic hazards in raw milk by metagenomics using Brucella as a model pathogen. Microb. Genom. 2021, 7, 000552. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, G.; Huang, D.W.; Wainwright, B.J. The mycobiome of Pocillopora acuta in Singapore. Coral Reefs 2021, 40, 1419–1427. [Google Scholar] [CrossRef]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, gix120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosius, J.; Dull, T.J.; Sleeter, D.D.; Noller, H.F. Gene organization and primary structure of a ribosomal RNA operon from Escherichia coli. J. Mol. Biol. 1981, 148, 107–127. [Google Scholar] [CrossRef]
- Zhang, X.; Tian, X.; Ma, L.; Feng, B.; Liu, Q.; Yuan, L.; Fan, C.; Huang, H.; Yang, Q. Biodiversity of the symbiotic bacteria associated with toxic marine dinoflagellate Alexandrium tamarense. J. Biosci. Med. 2015, 3, 23. [Google Scholar] [CrossRef] [Green Version]
- Lonsane, B.; Saucedo-Castaneda, G.; Raimbault, M.; Roussos, S.; Viniegra-Gonzalez, G.; Ghildyal, N.; Ramakrishna, M.; Krishnaiah, M.J.P.B. Scale-up strategies for solid state fermentation systems. Process. Biochem. 1992, 27, 259–273. [Google Scholar] [CrossRef]
- Yie, W.; Liu, X.; Lin, S.; Tan, J.; Pan, J.; Li, D.; Yang, H. The vertical distribution of bacterial and archaeal communities in the water and sediment of Lake Taihu. FEMS Microbiol. Ecol. 2009, 70, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginestet, C. ggplot2: Elegant Graphics for Data Analysis. J. R. Stat. Soc. Ser. A Stat. Soc. 2011, 174, 245. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.; O’Hara, R.; Simpson, G.; Solymos, P. vegan: Community Ecology Package. R Package Version 2.5-6, 2019. Available online: https://cran.r-project.org/web/packages/vegan (accessed on 10 May 2021).
- Chen, H.; Wang, M.; Zhang, H.; Wang, H.; Lv, Z.; Zhou, L.; Zhong, Z.; Lian, C.; Cao, L.; Li, C. An LRR-domain containing protein identified in Bathymodiolus platifrons serves as intracellular recognition receptor for the endosymbiotic methane-oxidation bacteria. Fish Shellfish Immunol. 2019, 93, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Marotz, C.A.; Sanders, J.G.; Zuniga, C.; Zaramela, L.S.; Knight, R.; Zengler, K. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome 2018, 6, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, M.; Wu, B.; Yan, G.; Li, G.; Sun, L.; Lu, G.; Zhou, W. Combined nanopore adaptive sequencing and enzyme-based host depletion efficiently enriched microbial sequences and identified missing respiratory pathogens. BMC Genomics 2021, 22, 732. [Google Scholar] [CrossRef]
- Zhang, C.; Cleveland, K.; Schnoll-Sussman, F.; McClure, B.; Bigg, M.; Thakkar, P.; Schultz, N.; Shah, M.A.; Betel, D. Identification of low abundance microbiome in clinical samples using whole genome sequencing. Genome Biol. 2015, 16, 265. [Google Scholar] [CrossRef] [Green Version]
- Zhilina, T.N.; Zavarzin, G.A.; Rainey, F.; Kevbrin, V.V.; Kostrikina, N.A.; Lysenko, A.M. Spirochaeta alkalica sp. nov., Spirochaeta africana sp. nov., and Spirochaeta asiatica sp. nov., alkaliphilic anaerobes from the Continental Soda Lakes in Central Asia and the East African Rift. Int. J. Syst. Bacteriol. 1996, 46, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Carella, F.; Aceto, S.; Pollaro, F.; Miccio, A.; Iaria, C.; Carrasco, N.; Prado, P.; De Vico, G. A mycobacterial disease is associated with the silent mass mortality of the pen shell Pinna nobilis along the Tyrrhenian coastline of Italy. Sci. Rep. 2019, 9, 2725. [Google Scholar] [CrossRef]
- Nowlan, J.P.; Lumsden, J.S.; Russell, S. Advancements in Characterizing Tenacibaculum Infections in Canada. Pathogens 2020, 9, 1029. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Method | Differential Enrichment Steps | DNA Extraction Kit |
|---|---|---|---|
| Perna canaliculus gill tissue homogenate | PS | No enrichment | DNeasy Power Soil Kit |
| PC | 1. Centrifugation a. 5 min at 500× g b. 10 min at 15,000× g 2. Final pellet washing with IBS | DNeasy Power Soil Kit | |
| PF | 1. Filtration a. 40 μm cell strainer b. 5 μm polycarbonate membrane 2. Centrifugation 5 min at 15,000× g 3. The final pellet was washed with IBS | DNeasy Power Soil Kit | |
| HZ | No enrichment | HostZero Microbial DNA Kit |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Chen, Y.; Zhang, S.; Lyu, Y.; Zou, Y.; Li, J. DNA Enrichment Methods for Microbial Symbionts in Marine Bivalves. Microorganisms 2022, 10, 393. https://doi.org/10.3390/microorganisms10020393
Li Q, Chen Y, Zhang S, Lyu Y, Zou Y, Li J. DNA Enrichment Methods for Microbial Symbionts in Marine Bivalves. Microorganisms. 2022; 10(2):393. https://doi.org/10.3390/microorganisms10020393
Chicago/Turabian StyleLi, Qiqi, Yu Chen, Si Zhang, Yuanjiao Lyu, Yiyang Zou, and Jie Li. 2022. "DNA Enrichment Methods for Microbial Symbionts in Marine Bivalves" Microorganisms 10, no. 2: 393. https://doi.org/10.3390/microorganisms10020393
APA StyleLi, Q., Chen, Y., Zhang, S., Lyu, Y., Zou, Y., & Li, J. (2022). DNA Enrichment Methods for Microbial Symbionts in Marine Bivalves. Microorganisms, 10(2), 393. https://doi.org/10.3390/microorganisms10020393