
The Leaf Microbiome of Tobacco Plants across Eight Chinese Provinces



Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Sample Treatment
2.3. 16S rDNA Gene Sequencing and Sequence Processing
2.4. Sequence Comparison against Reference Databases
2.5. Species Abundance Distribution (SAD) Fitting
2.6. Core Bacterial Community Determination
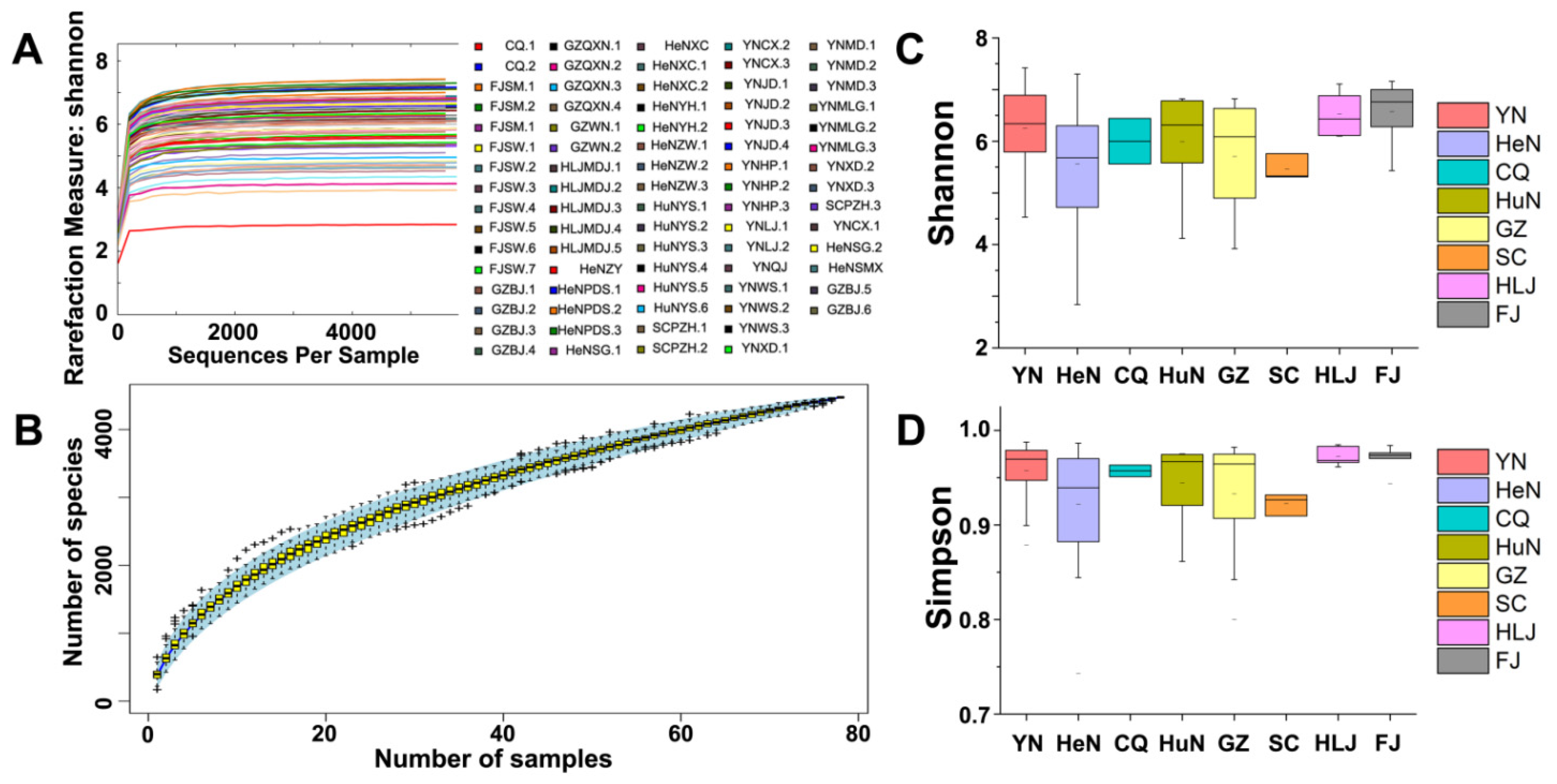
2.7. α-Diversity
2.8. β-Diversity
2.9. Redundancy Analysis (RDA)
2.10. Construction of Phylogenetic Tree and Taxonomic Tree
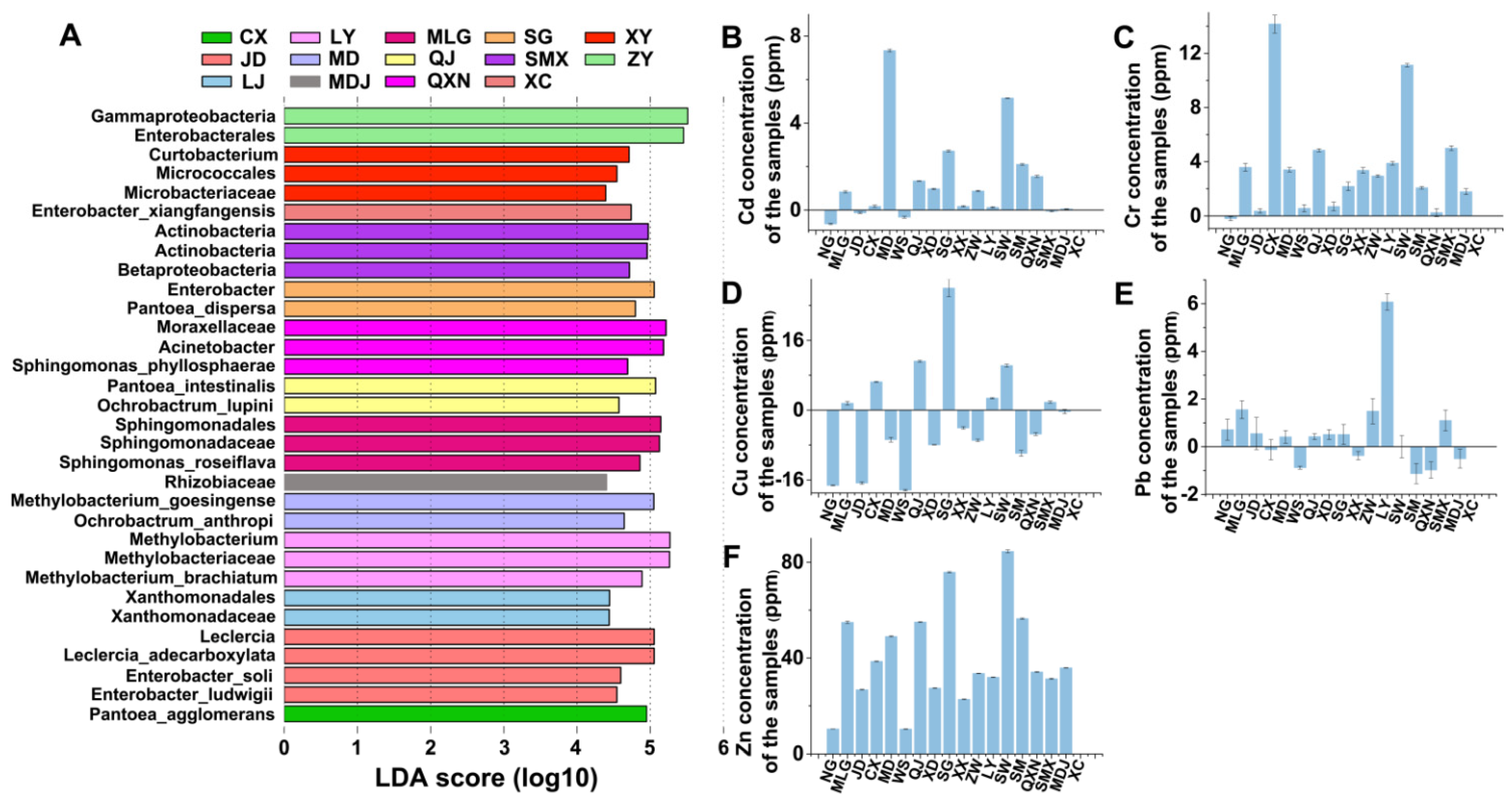
2.11. LEfSe Analysis
2.12. Functional Prediction with PICRUSt
2.13. Determination of Metals-Inductively Coupled Plasma Mass Spectrometry (ICP-MS)
2.14. Methods and Tools Used for Visualization
3. Results and Discussion
3.1. Species Abundance Distribution (SAD)
3.2. Functional Prediction with PICRUSt
3.3. α-Diversity Analysis of the Crop Samples
3.4. A National Core Bacterial Community

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Species | Function |
|---|---|---|
| Mastigocoleus | Mastigocoleus testarum | Function unknown |
| Atlantibacter | Atlantibacter hermannii | Function unknown |
| Salmonella | Salmonella enterica | Harmful for human body [38] |
| Leclercia | Leclercia adecarboxylata | Degradation of PAHs (pyrene) [45] |
| Enterobacter | Enterobacter soli | Degradation of indole-3-acetic acid and lignin [46,47] |
| Pantoea | Pantoea agglomerans | heavy metals reduction, nitrogen fixation, insecticidal activity [41,42,43] |
| Pseudomonas | Pseudomonas oryzihabitans Pseudomonas straminea | Insecticidal activity (root-knot nematode) [44] |
| Methylobacterium | Methylobacterium goesingense | Function unknown |
| Sphingomonas | Sphingomonas roseiflava Sphingomonas aurantiaca | Function unknown |
3.5. Provincial Core Bacterial Communities
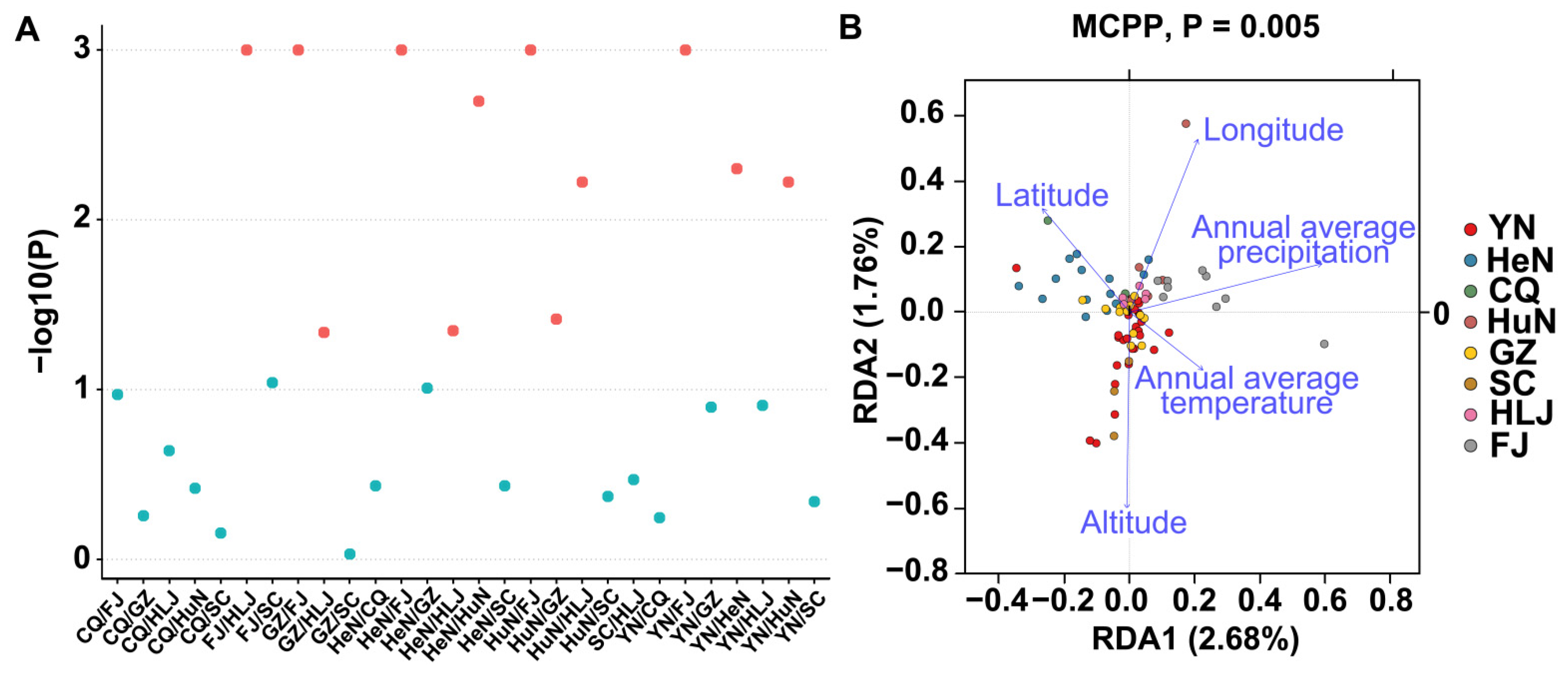
3.6. β-Diversity Analysis
3.7. Redundancy Analysis (RDA) and Correlation with Environmental Factors
3.8. Locally Specific Factors Drive Community Compositions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Alivisatos, A.P.; Blaser, M.J.; Brodie, E.L.; Chun, M.; Dangl, J.L.; Donohue, T.J.; Dorrestein, P.C.; Gilbert, J.A.; Green, J.L.; Jansson, J.K.; et al. A unified initiative to harness Earth’s microbiomes. Science 2015, 350, 507–508. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef] [Green Version]
- Bahram, M.; Hildebrand, F.; Forslund, S.K.; Anderson, J.L.; Soudzilovskaia, N.A.; Bodegom, P.M.; Bengtsson-Palme, J.; Anslan, S.; Coelho, L.P.; Harend, H.; et al. Structure and function of the global topsoil microbiome. Nature 2018, 560, 233–237. [Google Scholar] [CrossRef]
- Löffler, F.E.; Edwards, E.A. Harnessing microbial activities for environmental cleanup. Curr. Opin. Biotech. 2006, 17, 274–284. [Google Scholar] [CrossRef]
- Wu, L.; Ning, D.; Zhang, B.; Li, Y.; Zhang, P.; Shan, X.; Zhang, Q.; Brown, M.R.; Li, Z.; Van Nostrand, J.D.; et al. Global diversity and biogeography of bacterial communities in wastewater treatment plants. Nat. Microbiol. 2019, 4, 1183–1195. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Baquerizo, M.; Oliverio, A.M.; Brewer, T.E.; Benavent-González, A.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K.; Fierer, N. A global atlas of the dominant bacteria found in soil. Science 2018, 359, 320–325. [Google Scholar] [CrossRef] [Green Version]
- Ladha, J.K.; Barraquio, W.L.; Watanabe, I. Isolation and identification of nitrogen-fixing Enterobacter cloacae and Klebsiella planticola associated with rice plants. Can. J. Microbiol. 1983, 29, 1301–1308. [Google Scholar] [CrossRef]
- Ritpitakphong, U.; Falquet, L.; Vimoltust, A.; Berger, A.; Métraux, J.P.; L’Haridon, F. The microbiome of the leaf surface of Arabidopsis protects against a fungal pathogen. New Phytol. 2016, 210, 1033–1043. [Google Scholar] [CrossRef] [Green Version]
- Busby, P.E.; Peay, K.G.; Newcombe, G. Common foliar fungi of Populus trichocarpa modify Melampsora rust disease severity. New Phytol. 2015, 209, 1681–1692. [Google Scholar] [CrossRef] [Green Version]
- Leveau, J.H. A brief from the leaf: Latest research to inform our understanding of the phyllosphere microbiome. Curr. Opin. Microbiol. 2019, 49, 41–49. [Google Scholar] [CrossRef]
- English, C.F.; Bell, E.J.; Berger, A.J. Isolation of thermophiles from broadleaf tobacco and effect of pure culture inoculation on cigar aroma and mildness. Appl. Microbiol. 1967, 15, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Ruiz Gutierrez, V. Nicotine degradation by bacteria Enterobacter cloacae as nicotine degrader. Tobacco 1983, 22, 85–98. [Google Scholar]
- Zhao, M.; Cui, J. Analysis of bacterial communities on aging flue-cured tobacco leaves by 16S rDNA PCR-DGGE technology. Appl. Microbiol. Biotechnol. 2007, 73, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Liu, Y.; Li, F.; Wang, B.; Liu, G. Identification of dominant and fragrance-enhancing microorganisms of tobacco leaves during ripening. Acta Microbiol. Sin. 2009, 49, 624–630. [Google Scholar] [PubMed]
- Huang, J.; Yang, J.; Duan, Y.; Gu, W.; Gong, X.; Zhe, W.; Su, C.; Zhang, K.Q. Bacterial diversities on unaged and aging flue-cured tobacco leaves estimated by 16S rRNA sequence analysis. Appl. Microbiol. Biotechnol. 2010, 88, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Gu, W.; Zhe, W.; Zhang, K.Q.; Duan, Y.; Yang, J. Diversity and phylogeny of bacteria on Zimbabwe tobacco leaves estimated by 16S rRNA sequence analysis. Appl. Microbiol. Biotechnol. 2011, 92, 1033–1044. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, X.R.; Lei, L.P.; Xia, Z.Y. The diversity analysis of bacteria in curing tobacco leaves. Chin. J. Biol. Control 2009, 15, 20–24. [Google Scholar]
- Wu, X.-Y.; Liang, S.L.; Han, S.Y.; Chen, X.M.; Lin, Y. Diversity and phylogenetic analysis of bacterial communities on flue-cured tobacco leaves at different aged phases. Guangdong Agric. Sci. 2014, 9, 28–33. [Google Scholar]
- Zhao, H.; Li, S.G.; Sun, H.W.; Fan, L.I.; Tian, X.Y.; Yan, S.L.; Lu, X.B. Screening of dna extraction methods and pcr primers for microorganisms from maize leaves. Shandong Agricultural Sciences. Shandong Agric. Sci. 2012, 44, 9–14. [Google Scholar]
- Jin, H.; Li, B.L.; Jie, O.U.; Chen, L.M. Microbial population diversity of activated sludge for wastewater treatment. J. Microbiol. 2012, 32, 1–5. [Google Scholar]
- Lawley, B.; Tannock, G.W. Analysis of 16S rRNA Gene Amplicon Sequences Using the QIIME Software Package. Methods. Mol. Biol. 2017, 1537, 153–163. [Google Scholar] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGill, B.J.; Etienne, R.S.; Gray, J.S.; Alonso, D.; Anderson, M.J.; Benecha, H.K.; Dornelas, M.; Enquist, B.J.; Green, J.L.; He, F.; et al. Species abundance distributions: Moving beyond single prediction theories to integration within an ecological framework. Ecol. Lett. 2007, 10, 995–1015. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, W.R.; Locey, K.J.; Lennon, J.T. A macroecological theory of microbial biodiversity. Nat. Ecol. Evol. 2017, 1, 107. [Google Scholar] [CrossRef]
- Saunders, A.M.; Albertsen, M.; Vollertsen, J.; Nielsen, P.H. The activated sludge ecosystem contains a core community of abundant organisms. ISME J. 2016, 10, 11–20. [Google Scholar] [CrossRef]
- Gibson, D.J.; Ely, J.S.; Collins, S.L. The core-satellite species hypothesis provides a theoretical basis for Grime’s classification of dominant, subordinate, and transient species. J. Ecol. 1999, 87, 1064–1067. [Google Scholar] [CrossRef]
- Shannon, C.E. A mathematical theory of communication. Bell Labs Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Simpson, E.H. Measurement of diversity. Nature 1949, 163, 261. [Google Scholar] [CrossRef]
- Chao, A.; Chazdon, R.L.; Colwell, R.K.; Shen, T.J. A new statistical approach for assessing similarity of species composition with incidence and abundance data. Ecol. Lett. 2010, 8, 148–159. [Google Scholar] [CrossRef]
- Chao, A.; Yang, M. Stopping rules and estimation for recapture debugging with unequal failure rates. Biometrika 1993, 80, 193–201. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. Unifrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.T.; Peng, X.; Deng, G.H.; Sheng, H.F.; Yu, W.; Zhou, H.W.; Tam, N.F. Illumina sequencing of 16s rRNA tag revealed spatial variations of bacterial communities in a mangrove wetland. Microb. Ecol. 2013, 66, 96–104. [Google Scholar] [CrossRef] [PubMed]
- McArdle, B.H.; Anderson, M.J. Fitting multivariate models to community data: A comment on distanceâbased redundancy analysis. Ecology 2001, 82, 290–297. [Google Scholar] [CrossRef]
- Price, M.N.; De Hal, P.S.; Arkin, A.P. Fasttree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhowe, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, X.L.; Yang, H.; Geng, A.J.; Wen, D.; Wang, F.H. Determination of 15 trace and hazardous elements in edible laver and kelp by inductively coupled plasma mass spectrometry with wet digestion. Chin. J. Anal. Lab. 2015, 34, 803–806. [Google Scholar]
- Knodler, L.A.; Elfenbein, J.R. Salmonella enterica. Trends Microbiol. 2019, 27, 964–965. [Google Scholar] [CrossRef]
- Guida, B.S.; Garcia-Pichel, F. Draft genome assembly of a filamentous euendolithic (true boring) cyanobacterium, Mastigocoleus testarum strain BC008. Genome Announc. 2016, 4, e01574-15. [Google Scholar] [CrossRef] [Green Version]
- Hata, H.; Natori, T.; Mizuno, T.; Kanazawa, I.; Eldesouky, I.; Hayashi, M.; Miyata, M.; Fukunaga, H.; Ohji, S.; Hosoyama, A.; et al. Phylogenetics of family Enterobacteriaceae and proposal to reclassify Escherichia hermannii and Salmonella subterranea as Atlantibacter hermannii and Atlantibacter subterranea gen. nov., comb. nov. Microbiol. Immunol. 2016, 60, 303–311. [Google Scholar] [CrossRef]
- Feng, Y.J.; Shen, D.L.; Song, W. Rice endophyte Pantoea agglomerans YS19 promotes host plant growth and affects allocations of host photosynthates. J. Appl Microbiol 2010, 100, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Francis, C.A.; Obraztsova, A.Y.; Tebo, B.M. Dissimilatory metal reduction by the facultative anaerobe Pantoea agglomerans SP1. Appl. Environ. Microbiol. 2000, 66, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Nunes, C.; Usall, J.; Teixidó, N.; Viñas, I. Biological control of postharvest pear diseases using a bacterium, Pantoea agglomerans CPA-2. Int. J. Food Microbiol. 2001, 70, 53–61. [Google Scholar] [CrossRef]
- Vagelas, I.K.; Pembroke, B.; Gowen, S.R. The control of root-knot nematodes (Meloidogyne spp.) by Pseudomonas oryzihabitans and its immunological detection on tomato roots. Nematology 2007, 9, 363–370. [Google Scholar]
- Sarma, P.M.; Duraja, P.; Deshpande, S.; Lal, B. Degradation of pyrene by an enteric bacterium, Leclercia adecarboxylata PS4040. Biodegradation 2010, 21, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Daniel, K.M.; William, J.H.; Jorge, M.V. Enterobacter soli sp. nov.: A lignin-degrading γ-proteobacteria isolated from soil. Curr. Microbiol. 2011, 62, 1044–1049. [Google Scholar]
- Greenhut, I.V.; Slezak, B.L.; Leveau, J.H.J. iac gene expression in the indole-3-acetic acid-degrading soil bacterium Enterobacter soli LF7. Appl. Environ. Microbiol. 2018, 84, e01057-18. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.H.; Ren, Y.; Wu, C.F. The characteristics of aniline biodegradation by Ochrobactrum anthropi. Chin. J. Environ. Sci. 1998, 19, 22–24. [Google Scholar]
- Ozdemir, G.; Ozturk, T.; Ceyhan, N.; Isler, R.; Cosar, T. Heavy metal biosorption by biomass of Ochrobactrum anthropi producing exopolysaccharide in activated sludge. Bioresour. Technol. 2003, 90, 71–74. [Google Scholar] [CrossRef]
- Shi, X.Z.; Guo, R.J.; Takagi, K.; Miao, Z.Q.; Li, S.D. Chlorothalonil degradation by Ochrobactrum lupini strain TP-D1 and identification of its metabolites. World J. Microb. Biot. 2011, 27, 1755–1764. [Google Scholar] [CrossRef]
- Chen, S.; Hu, M.; Liu, J.; Zhong, G.; Yang, L.; Rizwan-ul-Haq, M.; Han, H. Biodegradation of beta-cypermethrin and 3-phenoxybenzoic acid by a novel Ochrobactrum lupini DG-S-01. J. Hazard Mater. 2011, 187, 433–440. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Biswas, D.; Sana, S.; Datta, S. Utilization of waste engine oil by Ochrobactrum pseudintermedium strain C1 that secretes an exopolysaccharide as a bioemulsifier. Biocatal. Agric. Biotechnol. 2014, 3, 167–176. [Google Scholar] [CrossRef]
- Ontañon, O.M.; González, P.S.; Agostini, E. Biochemical and molecular mechanisms involved in simultaneous phenol and Cr(VI) removal by Acinetobacter guillouiae SFC 500-1A. Environ. Sci. Pollut. Res. Int. 2015, 22, 13014–13023. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.B.; Han, L.R.; Feng, J.T.; Zhang, X. Screening of highly efficient cellulose Degradation Microbes and Construction of Composite Strains. J. Agric. Biotechnol. 2015, 23, 421–431. [Google Scholar]
- Zeng, X.M.; Huang, Q.Z. Identification on the pathogenic bacteria of a new tabacco disease-black rot. J. South China Agric. Univ. 1994, 15, 46–49. [Google Scholar]
- Thele, R.; Gumpert, H.; Christensen, L.B.; Worning, P.; Schønning, K.; Westh, H.; Hansen, T.A. Draft genome sequence of a Kluyvera intermedia isolate from a patient with a pancreatic abscess. J. Glob. Antimicrob. Resist. 2017, 10, 1–2. [Google Scholar] [CrossRef]
- Nicodemo, A.C.; Paez, J.I. Antimicrobial therapy for Stenotrophomonas maltophilia infections. Eur. J. Clin. Microbiol. Infect. Dis. 2007, 26, 229–237. [Google Scholar] [CrossRef]
- Zelazny, A.M.; Ding, L.; Goldberg, J.B.; Mijares, L.A.; Conlan, S.; Conville, P.S.; Stock, F.; Ballentine, S.J.; Olivier, K.N.; Sampaio, E.P.; et al. Adaptability and persistence of the emerging pathogen Bordetella petrii. PLoS ONE 2013, 8, e65102. [Google Scholar] [CrossRef] [Green Version]
- Kattar, M.M.; Chavez, J.F.; Limaye, A.P.; Rassoulian-Barrett, S.L.; Yarfitz, S.L.; Carlson, L.C.; Houze, Y.; Swanzy, S.; Wood, B.L.; Cookson, B.T. Application of 16S rRNA gene sequencing to identify Bordetella hinzii as the causative agent of fatal septicemia. J. Clin. Microbiol. 2000, 38, 789–794. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Wang, L.; Wang, W.; Yu, H.; Zhang, K.; Yao, Y.; Xu, P. Systematic unraveling of the unsolved pathway of nicotine degradation in Pseudomonas. PLoS Genet. 2013, 9, e1003923. [Google Scholar] [CrossRef] [Green Version]
- Fuhrman, J.A.; Steele, J.A.; Hewson, I.; Schwalbach, M.S.; Brown, M.V.; Green, J.L.; Brown, J.H. A latitudinal diversity gradient in planktonic marine bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 7774–7778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoebitz, M.; Ribaudo, C.M.; Pardo, M.A.; Cantore, M.L.; Curá, J.A. Plant growth promoting properties of a strain of enterobacter ludwigii isolated from lolium perenne rhizosphere. Soil Biol. Biochem. 2009, 41, 1768–1774. [Google Scholar] [CrossRef]
- Yousaf, S.; Afzal, M.; Reichenauer, T.G.; Brady, C.L.; Sessitsch, A. Hydrocarbon degradation, plant colonization and gene expression of alkane degradation genes by endophytic enterobacter ludwigii strains. Environ. Pollut. 2011, 159, 2675–2683. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.T.; Zhu, X.Z.; Chen, X.M.; Sun, Y.W.; Lv, B.T.; Wang, D.Q.; Wang, X. Colonization of functional endophytic Enterobacter sp. PRd5 to reduce pyrene contamination in crops. J. Nanjing Agric. Univ. 2020, 2, 274–283. [Google Scholar]
- Bahig, E.D. Plasmid Mediated Tolerance and Removal of Heavy Metals by Enterobacter sp. Am. J. Biochem. Biotechnol. 2009, 5, 47–53. [Google Scholar]
- Lu, L.; Chai, Q.; He, S.; Yang, C.; Zhang, D. Effects and mechanisms of phytoalexins on the removal of polycyclic aromatic hydrocarbons (pahs) by an endophytic bacterium isolated from ryegrass. Environ. Pollut. 2019, 253, 872–881. [Google Scholar] [CrossRef]
- Jeong, S.W.; Kim, H.K.; Yang, J.E.; Choi, Y.J. Removal of Pb(II) by Pellicle-like Biofilm-Producing Methylobacterium hispanicum EM2 Strain from Aqueous Media. Water 2019, 11, 2081. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.B. The study of bioremediation on heavy metal of cultured seawater by Sphingomonas sp. xj2 immobilized Sphingomonas strain. Adv. Mater. Res. 2011, 347–353, 1436–1441. [Google Scholar] [CrossRef]
- She, B.; Tao, X.; Huang, T.; Lu, G.; Zhou, Z.; Guo, C.; Dang, Z. Effects of nano bamboo charcoal on pahs-degrading strain Sphingomonas sp. gy2b. Ecotoxicol. Environ. Saf. 2016, 125, 35–42. [Google Scholar] [CrossRef]


| Sampling Location | Species | Function |
|---|---|---|
| Yunnan-Mangdui | Ochrobactrum anthropi | Adsorption of heavy metal ions [49] |
| Yunnan-Menglijiaojidi | Leclercia adecarboxylata | Degradation of PAHs [45] |
| Yunnan-Menglijiaojidi | Enterobacter ludwigii | Promotion of plant growth, alkane degradation [62,63] |
| Yunnan-Qujing | Ochrobactrum lupini | Degradation of pesticides (chlorothalonil and cypermethrin) [50,51] |
| Yunnan-Chuxiong | Pantoea agglomerans | Heavy metals detoxification [61] |
| Henan-Xuchang | Enterobacter xiangfangensis | Function unknown, but the corresponding genus is able to degrade PAHs and heavy metals [64,65] |
| Henan-Luoyang | Methylobacterium brachiatum | Function unknown, but the corresponding genus is able to degrade PAHs and heavy metals [66,67] |
| Guizhou-Qianxinan | Sphingomonas phyllosphaerae | Function unknown, but the corresponding genus is able to degrade PAHs and heavy metals [68,69] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, H.; Liu, Y.; Huang, Y.; Zhang, Z.; Tang, H. The Leaf Microbiome of Tobacco Plants across Eight Chinese Provinces. Microorganisms 2022, 10, 450. https://doi.org/10.3390/microorganisms10020450
Hu H, Liu Y, Huang Y, Zhang Z, Tang H. The Leaf Microbiome of Tobacco Plants across Eight Chinese Provinces. Microorganisms. 2022; 10(2):450. https://doi.org/10.3390/microorganisms10020450
Chicago/Turabian StyleHu, Haiyang, Yunli Liu, Yiqun Huang, Zhan Zhang, and Hongzhi Tang. 2022. "The Leaf Microbiome of Tobacco Plants across Eight Chinese Provinces" Microorganisms 10, no. 2: 450. https://doi.org/10.3390/microorganisms10020450
APA StyleHu, H., Liu, Y., Huang, Y., Zhang, Z., & Tang, H. (2022). The Leaf Microbiome of Tobacco Plants across Eight Chinese Provinces. Microorganisms, 10(2), 450. https://doi.org/10.3390/microorganisms10020450