
Mycobiome-Host Coevolution? The Mycobiome of Ancestral Human Populations Seems to Be Different and Less Diverse Than Those of Extant Native and Urban-Industrialized Populations

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
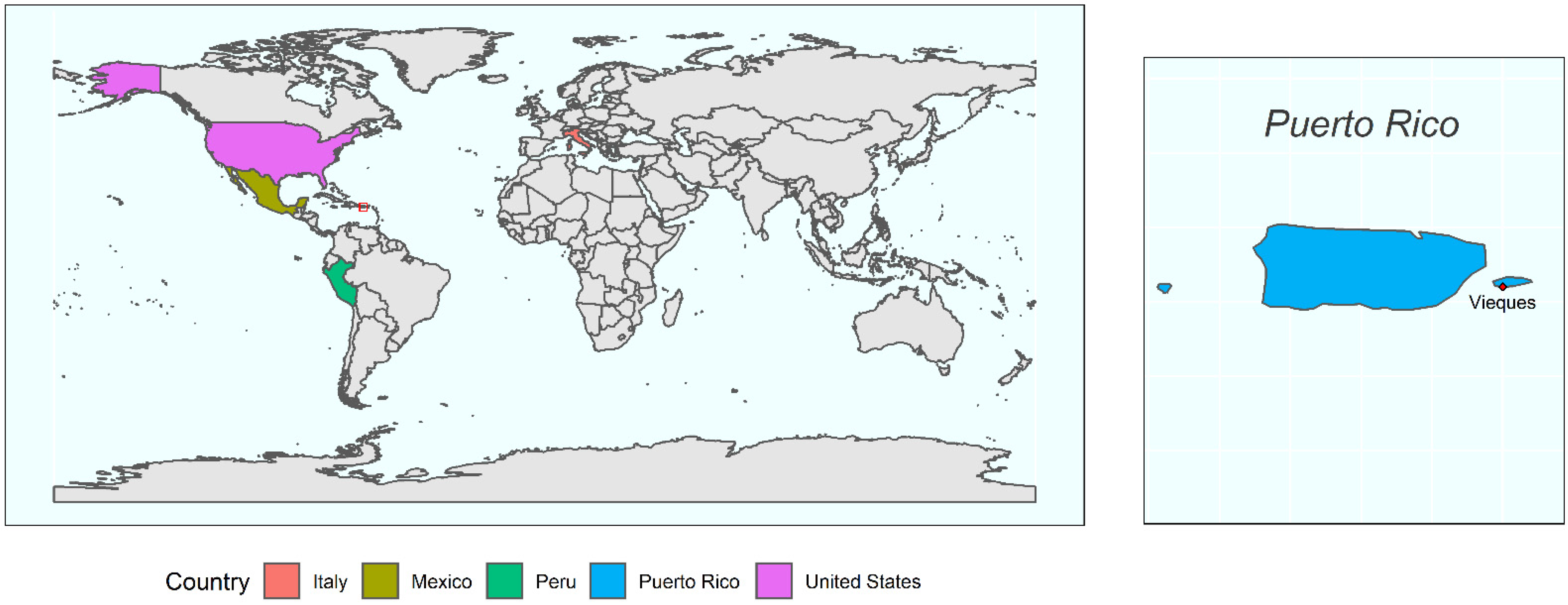
2.1. Study Site and Sample Collection
2.2. Microbial DNA Isolation and Contamination Control
2.3. Library Preparation and Shotgun Metagenomics Sequencing
2.4. Bioinformatics
2.4.1. Read Processing and Quality Control
2.4.2. Comparison to Other Samples
2.4.3. Taxonomic Profiling
2.4.4. Data and Statistical Analysis
3. Results
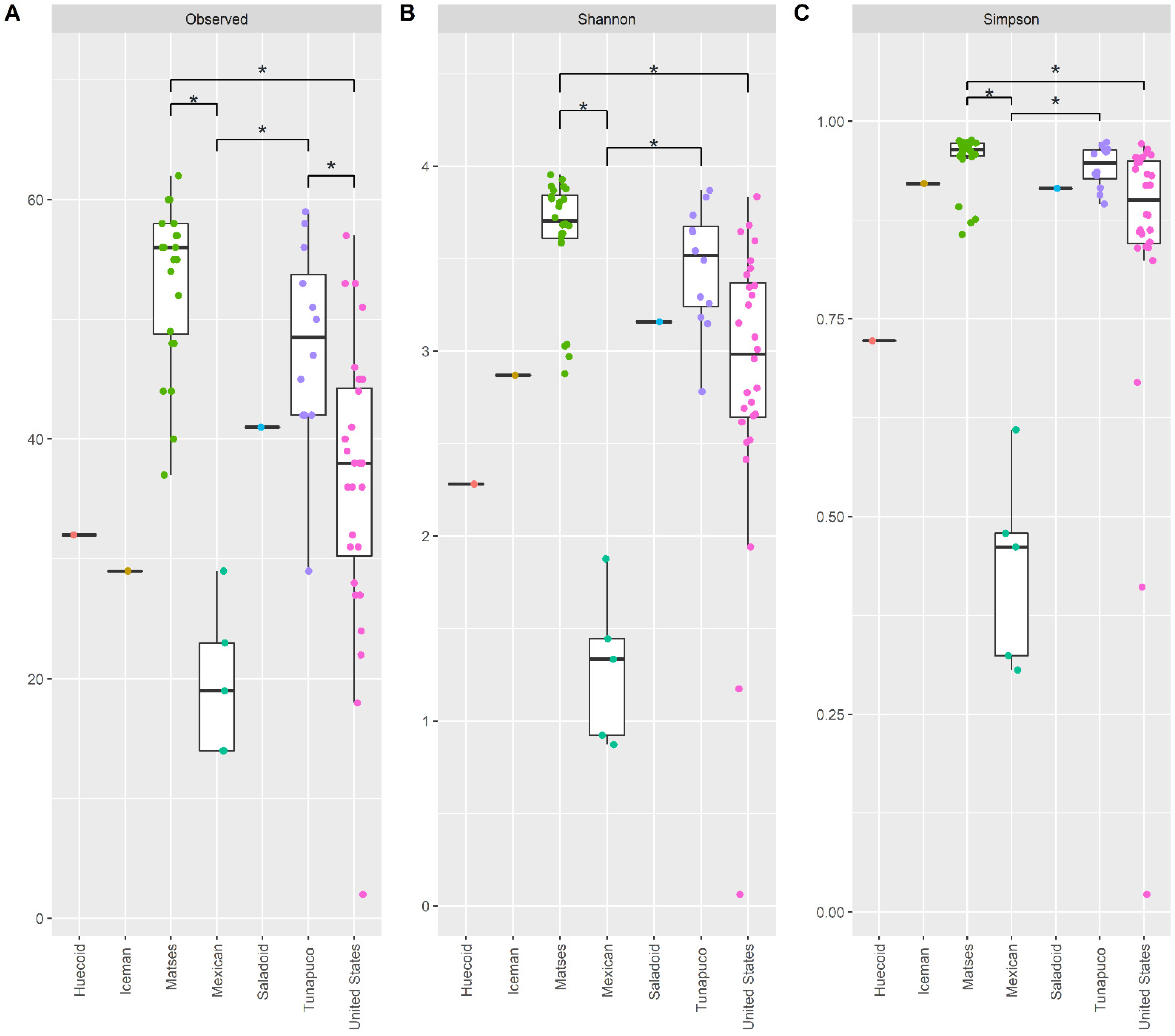
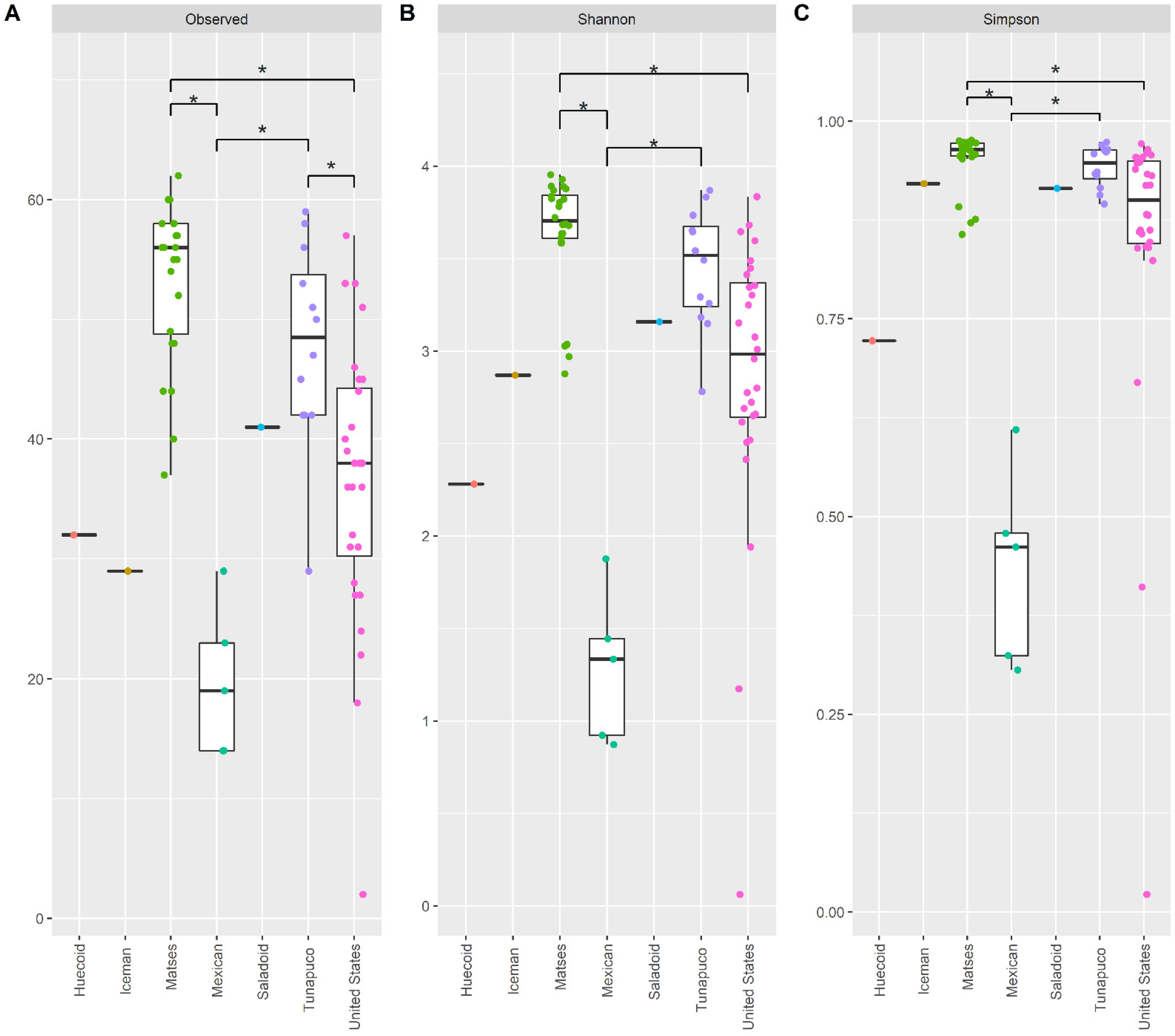
3.1. The Fecal Mycobiome of the Ancient Populations Is Less Diverse Than Those of Modern Populations
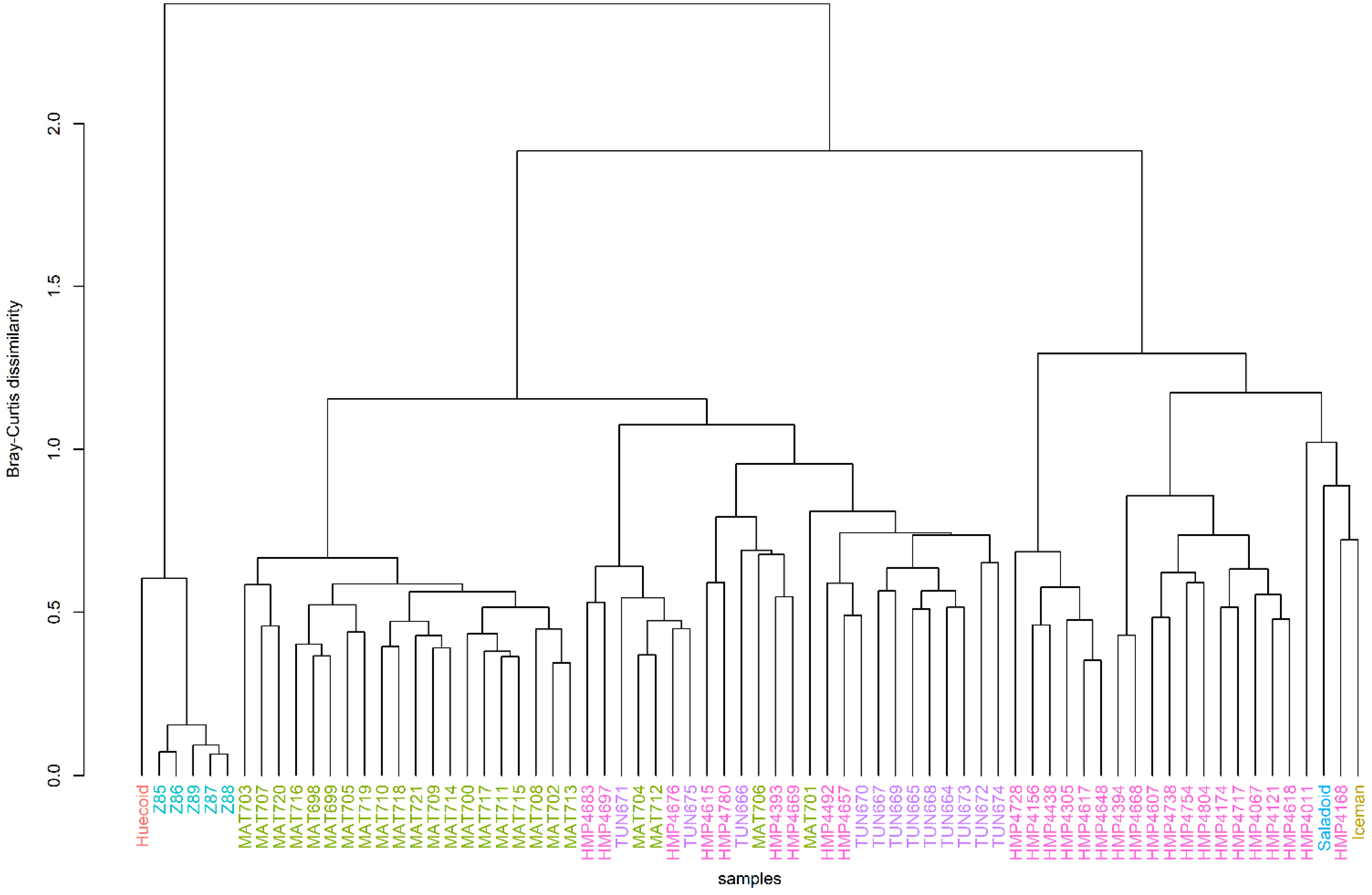
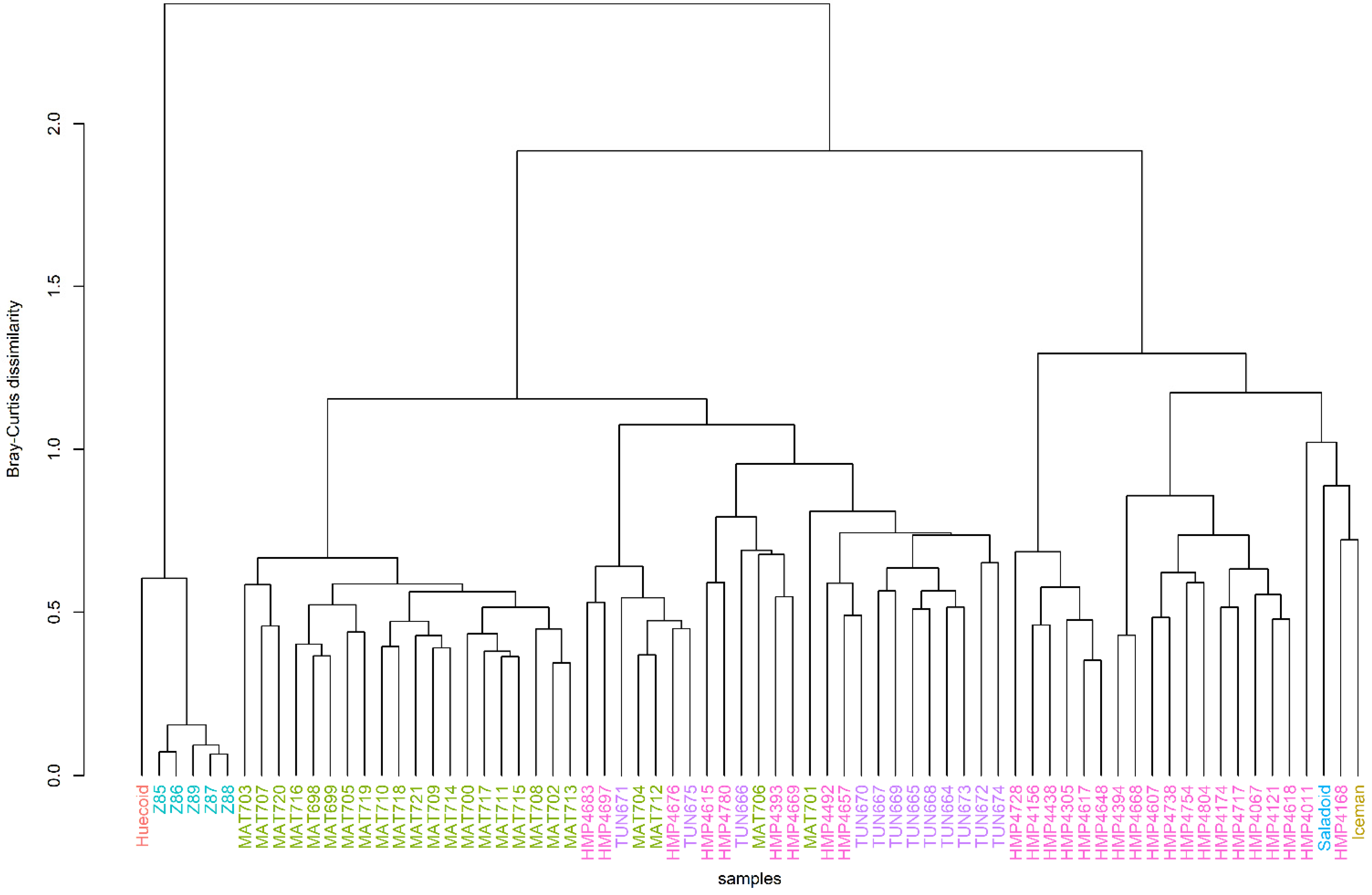
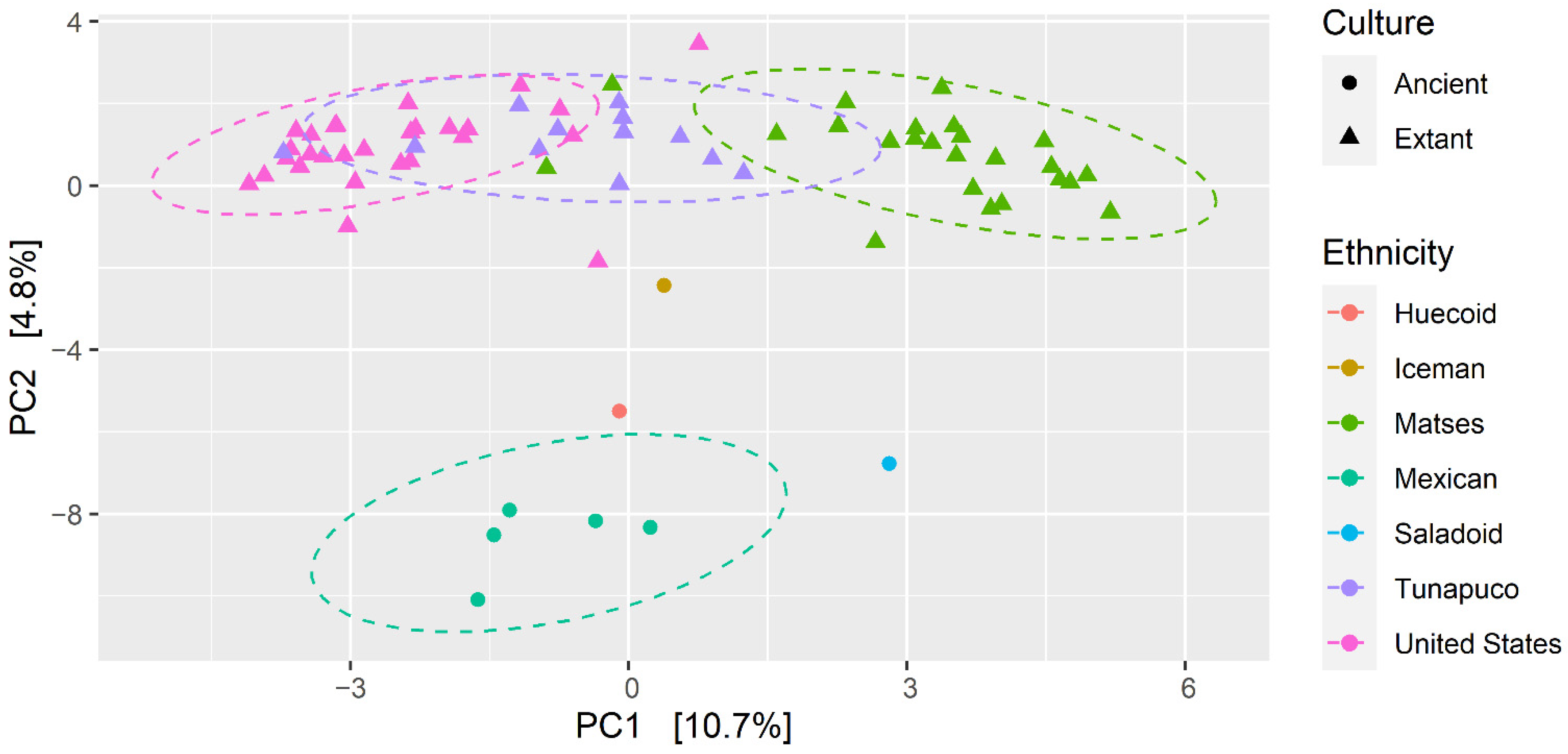
3.2. Hierarchical Clustering Revealed a Certain Degree of Clustering among the Ancient and Modern Populations
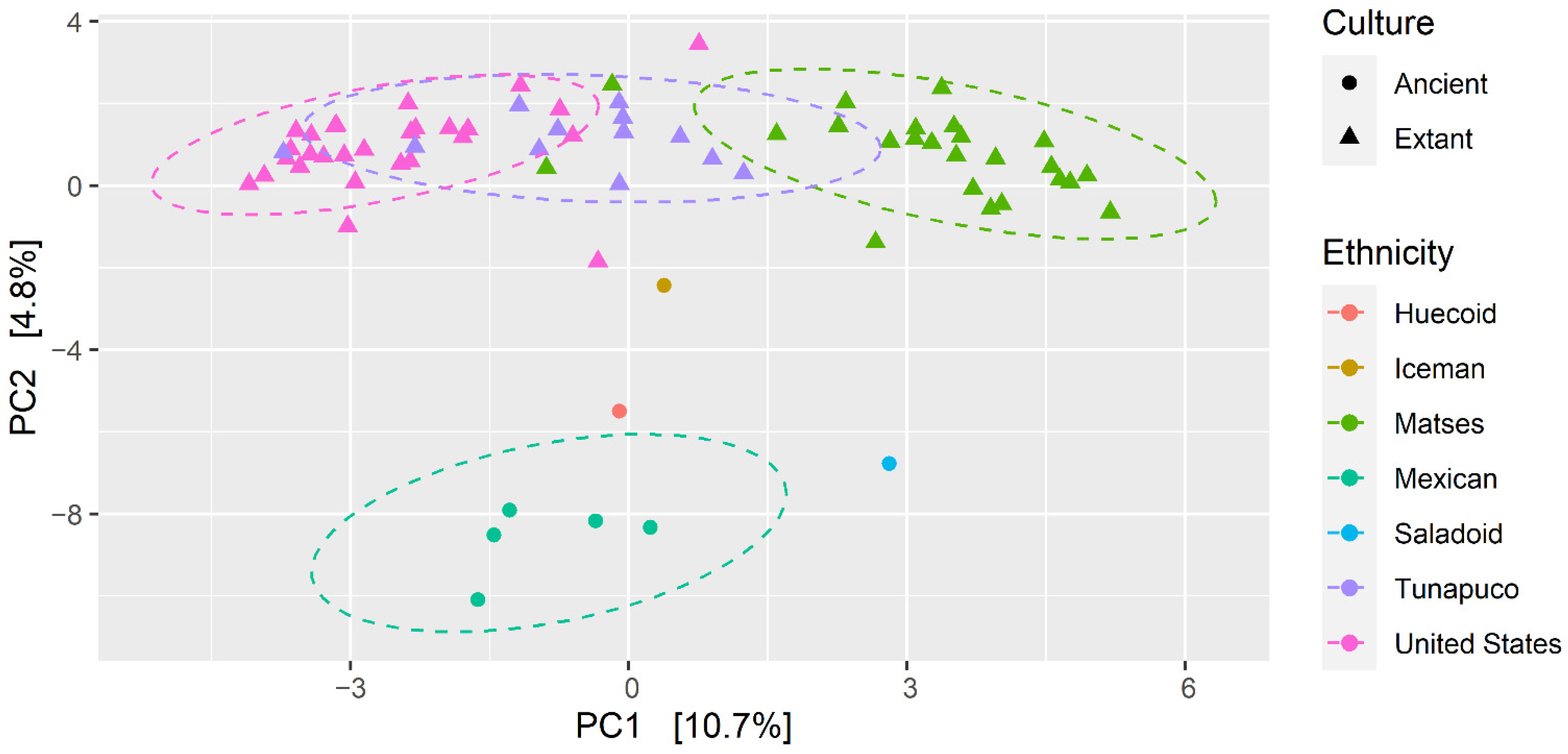
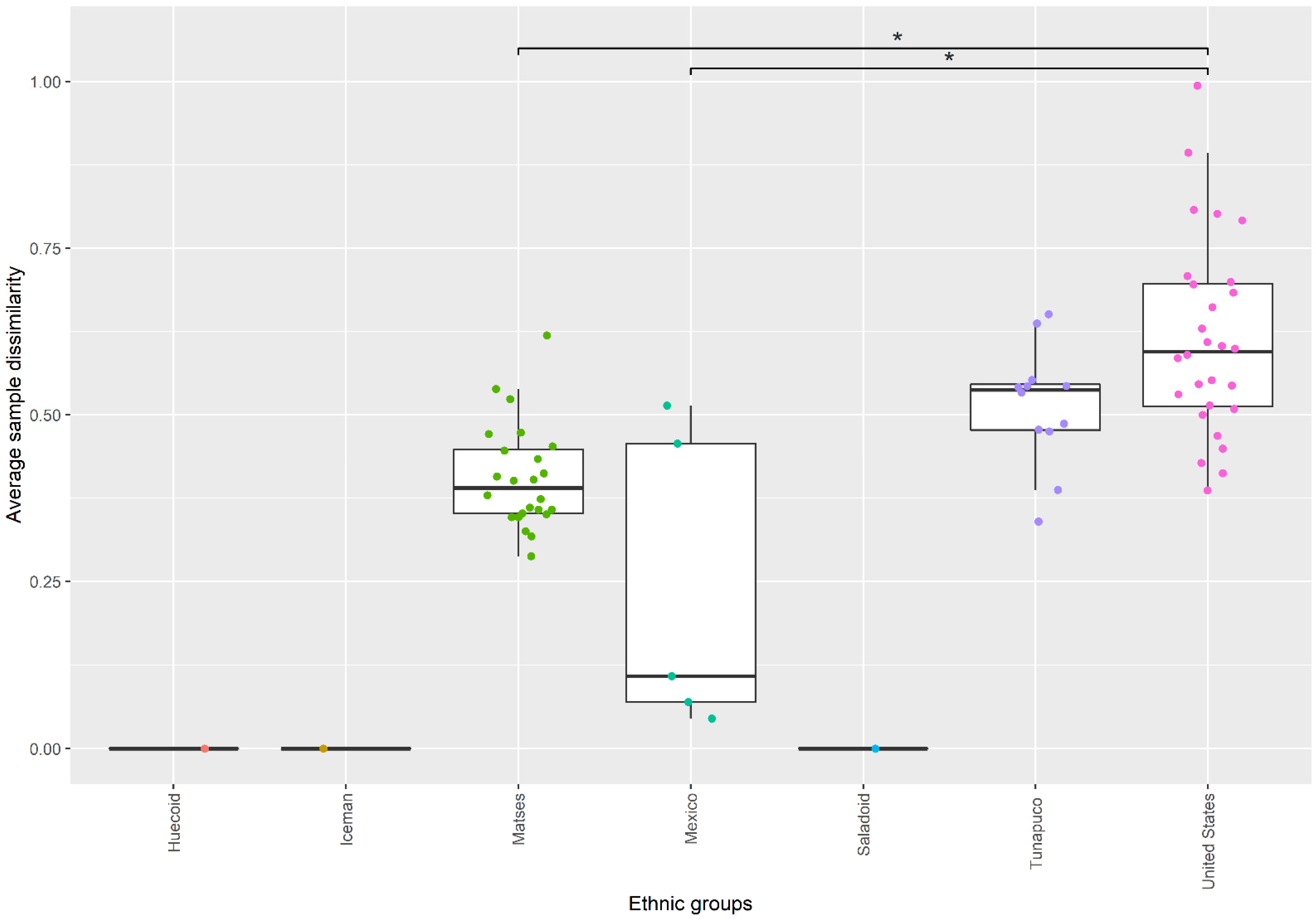
3.3. Fungal Communities of the Ancient Populations Differ from Those of Modern Populations
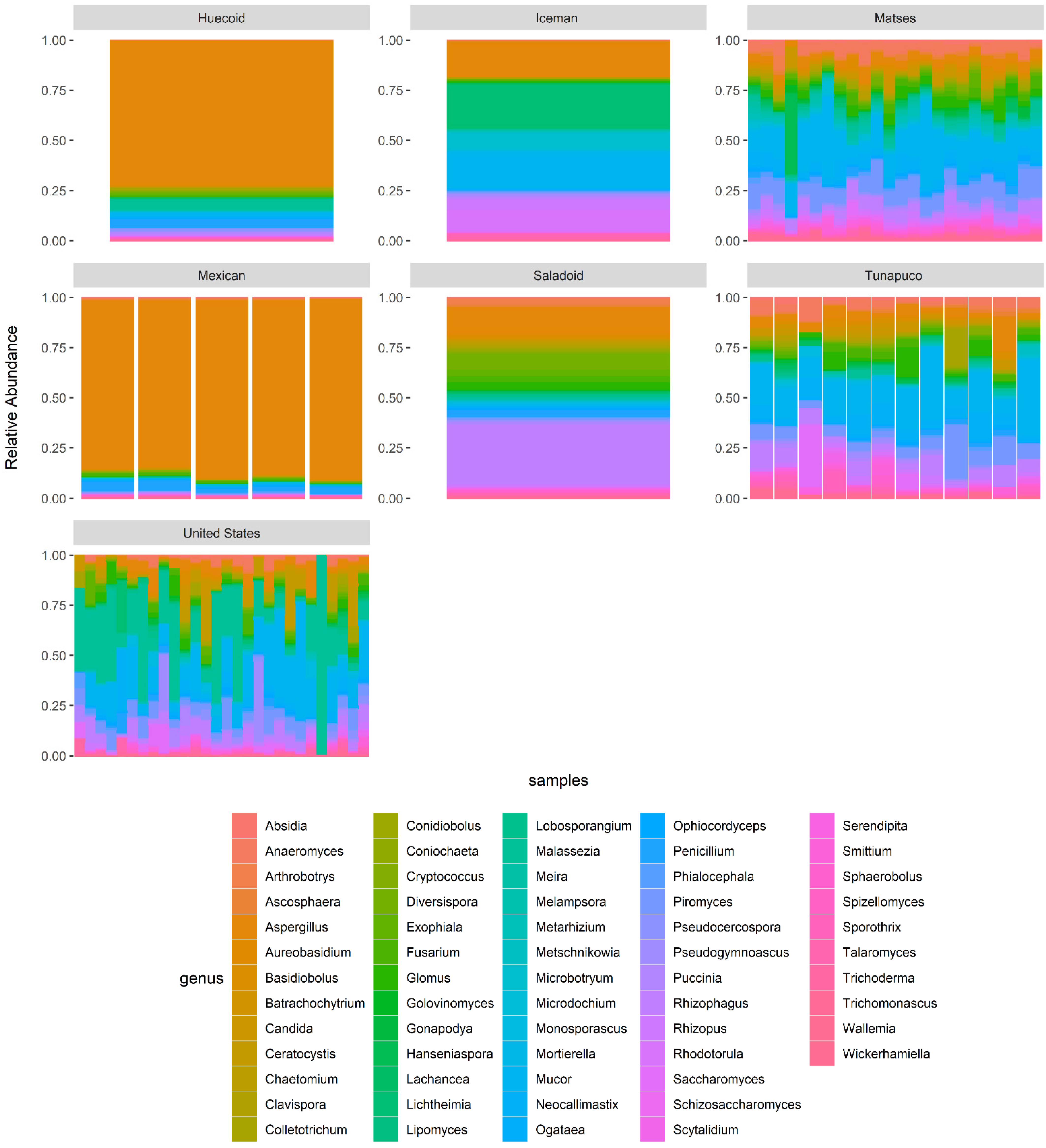
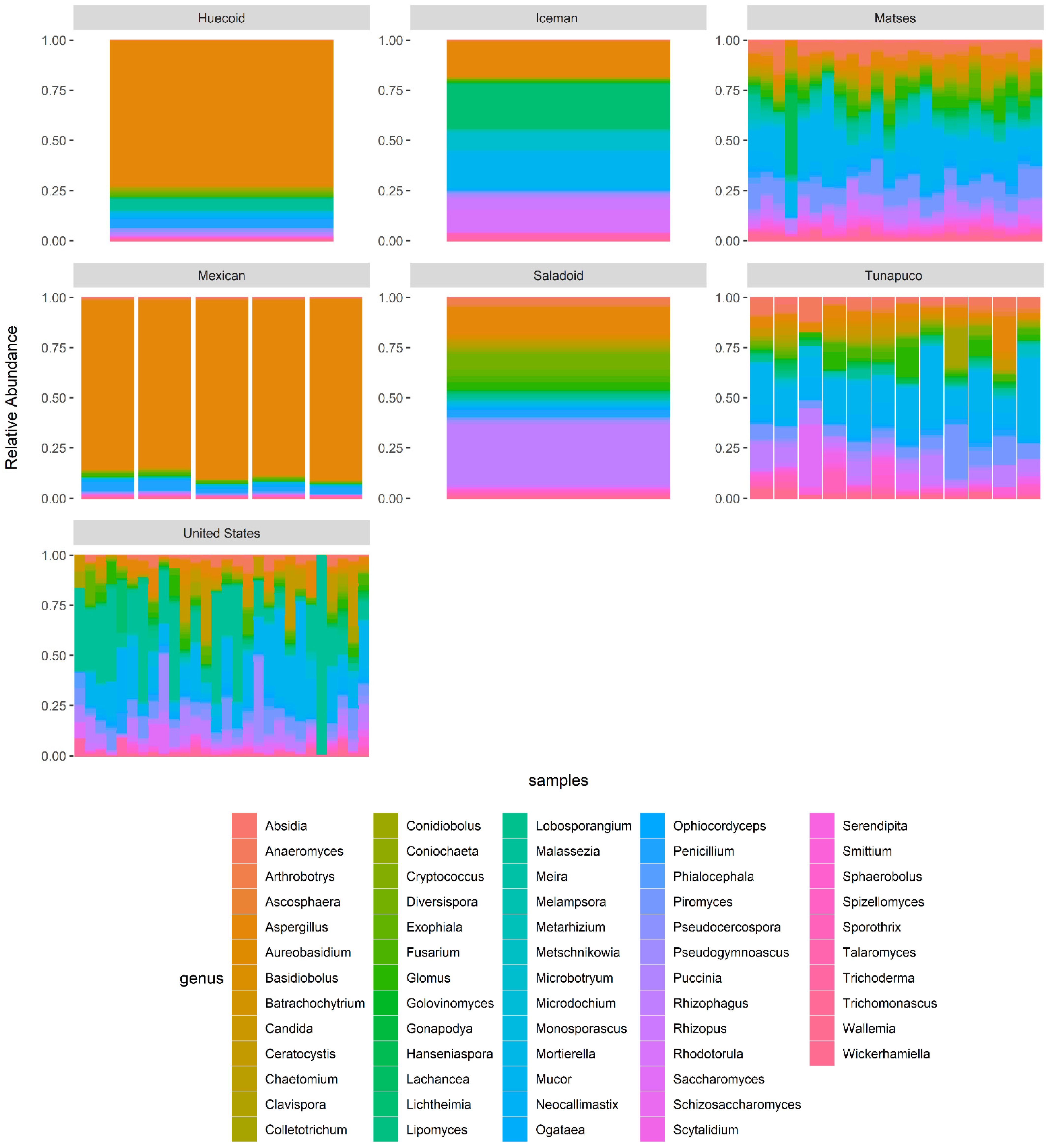
3.4. Metataxonomic Composition of the Samples Revealed Fungal Taxa That Differentiate Ancient and Modern Populations
3.5. Differentially Abundant Fungal Genera in the Ascomycota phylum Were the Main Drivers of the Differences between the Ancient and Modern Mycobiomes
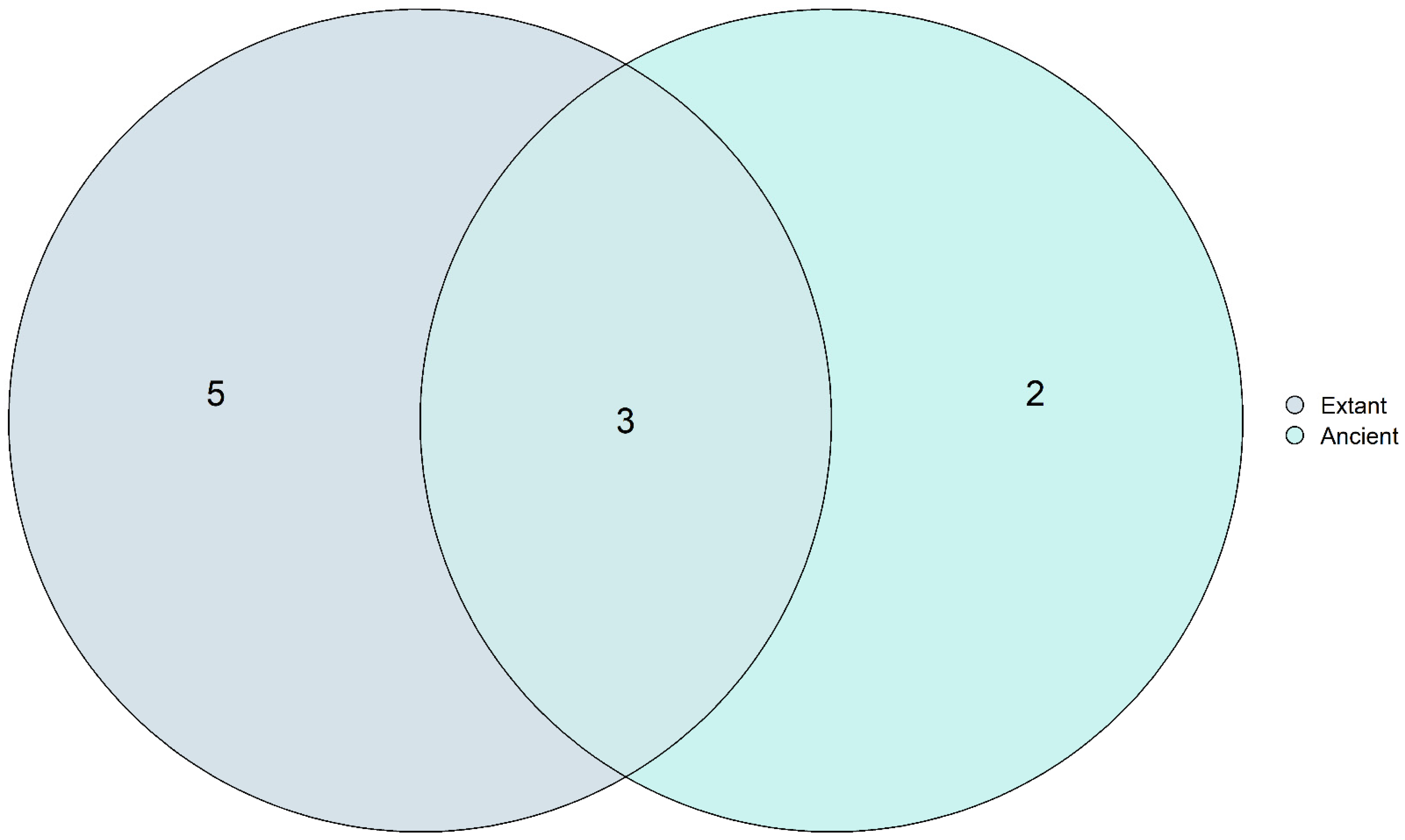
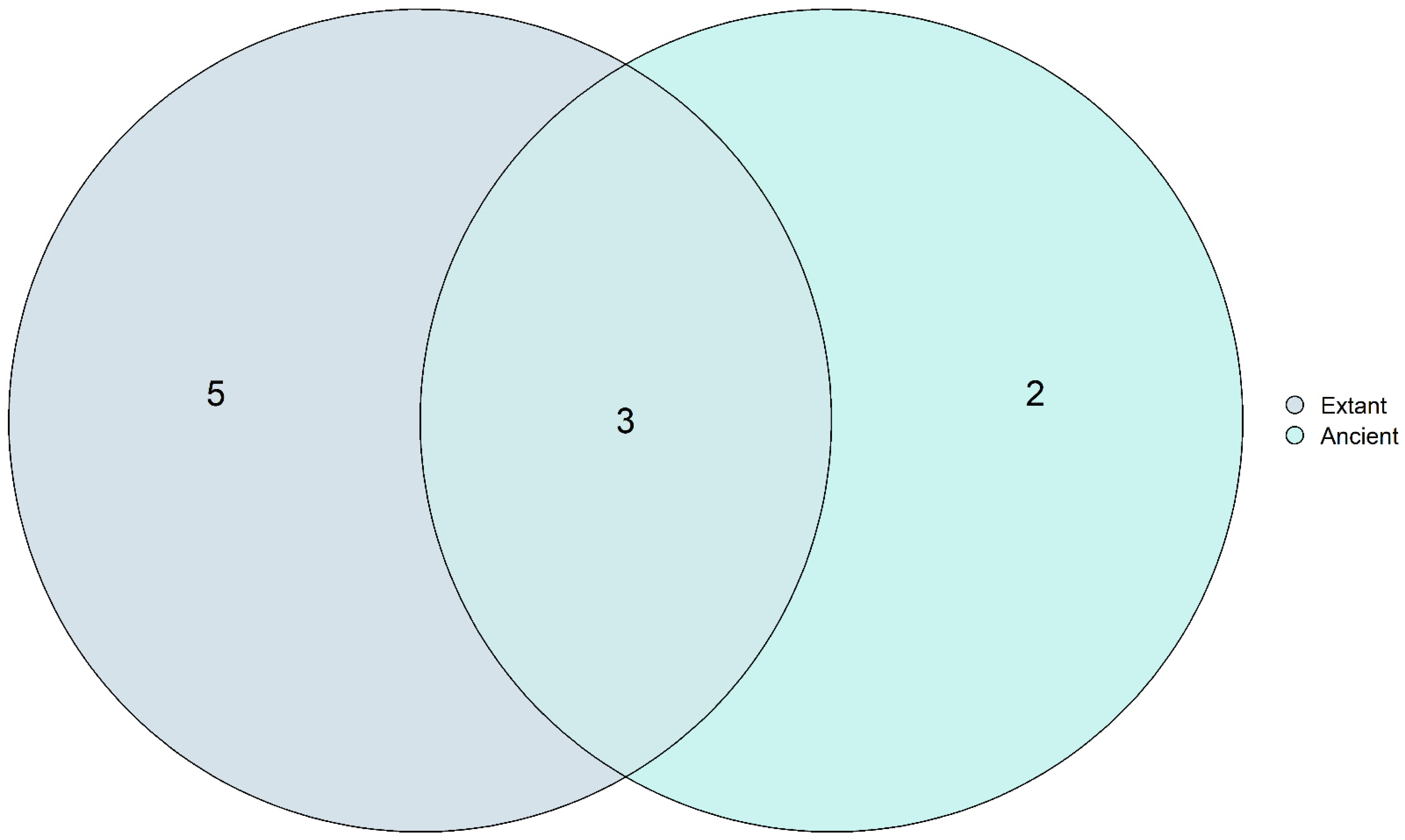
3.6. Core Mycobiome Results Show That the Most Abundant Fungal Genera Were Shared among the Ancient and Modern Populations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human Gut Microbiome Viewed across Age and Geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Deschasaux, M.; Bouter, K.E.; Prodan, A.; Levin, E.; Groen, A.K.; Herrema, H.; Tremaroli, V.; Bakker, G.J.; Attaye, I.; Pinto-Sietsma, S.-J.; et al. Depicting the Composition of Gut Microbiota in a Population with Varied Ethnic Origins but Shared Geography. Nat. Med. 2018, 24, 1526–1531. [Google Scholar] [CrossRef] [PubMed]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-Level Analysis of Gut Microbiome Variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef]
- He, Y.; Wu, W.; Zheng, H.-M.; Li, P.; McDonald, D.; Sheng, H.-F.; Chen, M.-X.; Chen, Z.-H.; Ji, G.-Y.; Zheng, Z.-D.-X.; et al. Regional Variation Limits Applications of Healthy Gut Microbiome Reference Ranges and Disease Models. Nat. Med. 2018, 24, 1532–1535. [Google Scholar] [CrossRef]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, T.; Kamm, M.A.; Colombel, J.-F.; Ng, S.C. Urbanization and the Gut Microbiota in Health and Inflammatory Bowel Disease. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 440–452. [Google Scholar] [CrossRef]
- Blaser, M.J. The Theory of Disappearing Microbiota and the Epidemics of Chronic Diseases. Nat. Rev. Immunol. 2017, 17, 461–463. [Google Scholar] [CrossRef] [PubMed]
- Vangay, P.; Johnson, A.J.; Ward, T.L.; Al-Ghalith, G.A.; Shields-Cutler, R.R.; Hillmann, B.M.; Lucas, S.K.; Beura, L.K.; Thompson, E.A.; Till, L.M.; et al. US Immigration Westernizes the Human Gut Microbiome. Cell 2018, 175, 962–972.e10. [Google Scholar] [CrossRef] [Green Version]
- Nash, A.K.; Auchtung, T.A.; Wong, M.C.; Smith, D.P.; Gesell, J.R.; Ross, M.C.; Stewart, C.J.; Metcalf, G.A.; Muzny, D.M.; Gibbs, R.A.; et al. The Gut Mycobiome of the Human Microbiome Project Healthy Cohort. Microbiome 2017, 5, 153. [Google Scholar] [CrossRef]
- Iliev, I.D.; Funari, V.A.; Taylor, K.D.; Nguyen, Q.; Reyes, C.N.; Strom, S.P.; Brown, J.; Becker, C.A.; Fleshner, P.R.; Dubinsky, M.; et al. Interactions between Commensal Fungi and the C-Type Lectin Receptor Dectin-1 Influence Colitis. Science 2012, 336, 1314–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliev, I.D.; Leonardi, I. Fungal Dysbiosis: Immunity and Interactions at Mucosal Barriers. Nat. Rev. Immunol. 2017, 17, 635–646. [Google Scholar] [CrossRef]
- Sokol, H.; Conway, K.L.; Zhang, M.; Choi, M.; Morin, B.; Cao, Z.; Villablanca, E.J.; Li, C.; Wijmenga, C.; Yun, S.H.; et al. Card9 Mediates Intestinal Epithelial Cell Restitution, T-Helper 17 Responses, and Control of Bacterial Infection in Mice. Gastroenterology 2013, 145, 591–601.e3. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Kamiya, T.; Liu, Y.; Kadoki, M.; Kakuta, S.; Oshima, K.; Hattori, M.; Takeshita, K.; Kanai, T.; Saijo, S.; et al. Inhibition of Dectin-1 Signaling Ameliorates Colitis by Inducing Lactobacillus-Mediated Regulatory T Cell Expansion in the Intestine. Cell Host Microbe 2015, 18, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Pan, D.; Zhou, Z.; You, Y.; Jiang, C.; Zhao, X.; Lin, X. Dectin-3 Deficiency Promotes Colitis Development Due to Impaired Antifungal Innate Immune Responses in the Gut. PLoS Pathog. 2016, 12, e1005662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tito, R.Y.; Knights, D.; Metcalf, J.; Obregon-Tito, A.J.; Cleeland, L.; Najar, F.; Roe, B.; Reinhard, K.; Sobolik, K.; Belknap, S.; et al. Insights from Characterizing Extinct Human Gut Microbiomes. PLoS ONE 2012, 7, e51146. [Google Scholar] [CrossRef]
- Tito, R.Y.; Macmil, S.; Wiley, G.; Najar, F.; Cleeland, L.; Qu, C.; Wang, P.; Romagne, F.; Leonard, S.; Ruiz, A.J.; et al. Phylotyping and Functional Analysis of Two Ancient Human Microbiomes. PLoS ONE 2008, 3, e3703. [Google Scholar] [CrossRef]
- Cano, R.J.; Rivera-Perez, J.; Toranzos, G.A.; Santiago-Rodriguez, T.M.; Narganes-Storde, Y.M.; Chanlatte-Baik, L.; García-Roldán, E.; Bunkley-Williams, L.; Massey, S.E. Paleomicrobiology: Revealing Fecal Microbiomes of Ancient Indigenous Cultures. PLoS ONE 2014, 9, e106833. [Google Scholar] [CrossRef]
- Santiago-Rodriguez, T.M.; Narganes-Storde, Y.M.; Chanlatte, L.; Crespo-Torres, E.; Toranzos, G.A.; Jimenez-Flores, R.; Hamrick, A.; Cano, R.J. Microbial Communities in Pre-Columbian Coprolites. PLoS ONE 2013, 8, e65191. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Perez, J.I.; Cano, R.J.; Narganes-Storde, Y.; Chanlatte-Baik, L.; Toranzos, G.A. Retroviral DNA Sequences as a Means for Determining Ancient Diets. PLoS ONE 2015, 10, e0144951. [Google Scholar] [CrossRef] [Green Version]
- Appelt, S.; Fancello, L.; Le Bailly, M.; Raoult, D.; Drancourt, M.; Desnues, C. Viruses in a 14th-Century Coprolite. Appl. Environ. Microbiol. 2014, 80, 2648–2655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiscovitch-Russo, R.; Rivera-Perez, J.; Narganes-Storde, Y.M.; García-Roldán, E.; Bunkley-Williams, L.; Cano, R.; Toranzos, G.A. Pre-Columbian Zoonotic Enteric Parasites: An Insight into Puerto Rican Indigenous Culture Diets and Life Styles. PLoS ONE 2020, 15, e0227810. [Google Scholar] [CrossRef] [PubMed]
- Chanlatte Baik, L.; Narganes-Sorde, Y. Cultura La Hueca, 1st ed.; Museo de Historia, Antropologia y Arte, Universidad de Puerto Rico: Recinto de Rio Piedras, PR, USA, 2005; pp. 1–6. [Google Scholar]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A Flexible Pipeline for Genome-Resolved Metagenomic Data Analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babraham Bioinformatics—FastQC A Quality Control Tool for High throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 10 December 2020).
- Tett, A.; Huang, K.D.; Asnicar, F.; Fehlner-Peach, H.; Pasolli, E.; Karcher, N.; Armanini, F.; Manghi, P.; Bonham, K.; Zolfo, M.; et al. The Prevotella Copri Complex Comprises Four Distinct Clades Underrepresented in Westernized Populations. Cell Host Microbe 2019, 26, 666–679.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obregon-Tito, A.J.; Tito, R.Y.; Metcalf, J.; Sankaranarayanan, K.; Clemente, J.C.; Ursell, L.K.; Zech Xu, Z.; Van Treuren, W.; Knight, R.; Gaffney, P.M.; et al. Subsistence Strategies in Traditional Societies Distinguish Gut Microbiomes. Nat. Commun. 2015, 6, 6505. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Price, J.; Mahurkar, A.; Rahnavard, G.; Crabtree, J.; Orvis, J.; Hall, A.B.; Brady, A.; Creasy, H.H.; McCracken, C.; Giglio, M.G.; et al. Strains, Functions and Dynamics in the Expanded Human Microbiome Project. Nature 2017, 550, 61–66. [Google Scholar] [CrossRef]
- Brooks, R.H.; Kaplan, L.; Cutler, H.C.; Whitaker, T.W. Plant Material from a Cave on the Rio Zape, Durango, Mexico. Am. Antiq. 1962, 27, 356–369. [Google Scholar] [CrossRef]
- Morrow, J.J.; Newby, J.; Piombino-Mascali, D.; Reinhard, K.J. Taphonomic Considerations for the Analysis of Parasites in Archaeological Materials. Int. J. Paleopathol. 2016, 13, 56–64. [Google Scholar] [CrossRef]
- Haak, W.; Lazaridis, I.; Patterson, N.; Rohland, N.; Mallick, S.; Llamas, B.; Brandt, G.; Nordenfelt, S.; Harney, E.; Stewardson, K.; et al. Massive Migration from the Steppe Was a Source for Indo-European Languages in Europe. Nature 2015, 522, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Keller, A.; Graefen, A.; Ball, M.; Matzas, M.; Boisguerin, V.; Maixner, F.; Leidinger, P.; Backes, C.; Khairat, R.; Forster, M.; et al. New Insights into the Tyrolean Iceman’s Origin and Phenotype as Inferred by Whole-Genome Sequencing. Nat. Commun. 2012, 3, 698. [Google Scholar] [CrossRef] [Green Version]
- Müller, W.; Fricke, H.; Halliday, A.N.; McCulloch, M.T.; Wartho, J.-A. Origin and Migration of the Alpine Iceman. Science 2003, 302, 862–866. [Google Scholar] [CrossRef] [Green Version]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and Sensitive Taxonomic Classification for Metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast Metagenomic Sequence Classification Using Exact Alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [Green Version]
- Rosa, P.S.L.; Brooks, J.P.; Deych, E.; Boone, E.L.; Edwards, D.J.; Wang, Q.; Sodergren, E.; Weinstock, G.; Shannon, W.D. Hypothesis Testing and Power Calculations for Taxonomic-Based Human Microbiome Data. PLoS ONE 2012, 7, e52078. [Google Scholar] [CrossRef] [Green Version]
- Magurran, A.E. Ecological Diversity and Its Measurement; Princeton University Press: Princeton, NJ, USA, 1988; ISBN 978-0-691-08491-6. [Google Scholar]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McArdle, B.H.; Anderson, M.J. Fitting Multivariate Models to Community Data: A Comment on Distance-Based Redundancy Analysis. Ecology 2001, 82, 290–297. [Google Scholar] [CrossRef]
- Aitchison, J. The Statistical Analysis of Compositional Data; Chapman and Hall: London, UK; New York, NY, USA, 1986; ISBN 978-0-412-28060-3. [Google Scholar]
- Dixon, P. VEGAN, A Package of R Functions for Community Ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Kabwe, M.H.; Vikram, S.; Mulaudzi, K.; Jansson, J.K.; Makhalanyane, T.P. The Gut Mycobiota of Rural and Urban Individuals Is Shaped by Geography. BMC Microbiol. 2020, 20, 257. [Google Scholar] [CrossRef]
- Sun, Y.; Zuo, T.; Cheung, C.P.; Gu, W.; Wan, Y.; Zhang, F.; Chen, N.; Zhan, H.; Yeoh, Y.K.; Niu, J.; et al. Population-Level Configurations of Gut Mycobiome Across 6 Ethnicities in Urban and Rural China. Gastroenterology 2021, 160, 272–286.e11. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of Diet in Shaping Gut Microbiota Revealed by a Comparative Study in Children from Europe and Rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [Green Version]
- De Filippo, C.; Di Paola, M.; Ramazzotti, M.; Albanese, D.; Pieraccini, G.; Banci, E.; Miglietta, F.; Cavalieri, D.; Lionetti, P. Diet, Environments, and Gut Microbiota. A Preliminary Investigation in Children Living in Rural and Urban Burkina Faso and Italy. Front. Microbiol. 2017, 8, 1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnorr, S.L.; Candela, M.; Rampelli, S.; Centanni, M.; Consolandi, C.; Basaglia, G.; Turroni, S.; Biagi, E.; Peano, C.; Severgnini, M.; et al. Gut Microbiome of the Hadza Hunter-Gatherers. Nat. Commun. 2014, 5, 3654. [Google Scholar] [CrossRef] [PubMed]
- Clemente, J.C.; Pehrsson, E.C.; Blaser, M.J.; Sandhu, K.; Gao, Z.; Wang, B.; Magris, M.; Hidalgo, G.; Contreras, M.; Noya-Alarcón, Ó.; et al. The Microbiome of Uncontacted Amerindians. Sci. Adv. 2015, 1, e1500183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, I.; Stegen, J.C.; Maldonado-Gómez, M.X.; Eren, A.M.; Siba, P.M.; Greenhill, A.R.; Walter, J. The Gut Microbiota of Rural Papua New Guineans: Composition, Diversity Patterns, and Ecological Processes. Cell Rep. 2015, 11, 527–538. [Google Scholar] [CrossRef] [Green Version]
- Rampelli, S.; Schnorr, S.L.; Consolandi, C.; Turroni, S.; Severgnini, M.; Peano, C.; Brigidi, P.; Crittenden, A.N.; Henry, A.G.; Candela, M. Metagenome Sequencing of the Hadza Hunter-Gatherer Gut Microbiota. Curr. Biol. CB 2015, 25, 1682–1693. [Google Scholar] [CrossRef] [Green Version]
- Gomez, A.; Petrzelkova, K.J.; Burns, M.B.; Yeoman, C.J.; Amato, K.R.; Vlckova, K.; Modry, D.; Todd, A.; Jost Robinson, C.A.; Remis, M.J.; et al. Gut Microbiome of Coexisting BaAka Pygmies and Bantu Reflects Gradients of Traditional Subsistence Patterns. Cell Rep. 2016, 14, 2142–2153. [Google Scholar] [CrossRef] [Green Version]
- Mancabelli, L.; Milani, C.; Lugli, G.A.; Turroni, F.; Ferrario, C.; van Sinderen, D.; Ventura, M. Meta-Analysis of the Human Gut Microbiome from Urbanized and Pre-Agricultural Populations. Environ. Microbiol. 2017, 19, 1379–1390. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Quinque, D.; Horz, H.-P.; Li, M.; Rzhetskaya, M.; Raff, J.A.; Hayes, M.G.; Stoneking, M. Comparative Analysis of the Human Saliva Microbiome from Different Climate Zones: Alaska, Germany, and Africa. BMC Microbiol. 2014, 14, 316. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.K.; Paul, S.; Dutta, C. Geography, Ethnicity or Subsistence-Specific Variations in Human Microbiome Composition and Diversity. Front. Microbiol. 2017, 8, 1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinöcker, M.K.; Lindseth, I.A. The Western Diet-Microbiome-Host Interaction and Its Role in Metabolic Disease. Nutrients 2018, 10, 365. [Google Scholar] [CrossRef] [Green Version]
- Huseyin, C.E.; Rubio, R.C.; O’Sullivan, O.; Cotter, P.D.; Scanlan, P.D. The Fungal Frontier: A Comparative Analysis of Methods Used in the Study of the Human Gut Mycobiome. Front. Microbiol. 2017, 8, 1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, M.L.; Limon, J.J.; Underhill, D.M. Immunity to Commensal Fungi: Detente and Disease. Annu. Rev. Pathol. 2017, 12, 359–385. [Google Scholar] [CrossRef]
- Goodley, J.M.; Clayton, Y.M.; Hay, R.J. Environmental Sampling for Aspergilli during Building Construction on a Hospital Site. J. Hosp. Infect. 1994, 26, 27–35. [Google Scholar] [CrossRef]
- Li, J.; Chen, D.; Yu, B.; He, J.; Zheng, P.; Mao, X.; Yu, J.; Luo, J.; Tian, G.; Huang, Z.; et al. Fungi in Gastrointestinal Tracts of Human and Mice: From Community to Functions. Microb. Ecol. 2018, 75, 821–829. [Google Scholar] [CrossRef]
- Li, Q.; Wang, C.; Tang, C.; He, Q.; Li, N.; Li, J. Dysbiosis of Gut Fungal Microbiota Is Associated with Mucosal Inflammation in Crohn’s Disease. J. Clin. Gastroenterol. 2014, 48, 513–523. [Google Scholar] [CrossRef] [Green Version]
- Gouba, N.; Raoult, D.; Drancourt, M. Eukaryote Culturomics of the Gut Reveals New Species. PLoS ONE 2014, 9, e106994. [Google Scholar] [CrossRef] [PubMed]
- Ukhanova, M.; Wang, X.; Baer, D.J.; Novotny, J.A.; Fredborg, M.; Mai, V. Effects of Almond and Pistachio Consumption on Gut Microbiota Composition in a Randomised Cross-over Human Feeding Study. Br. J. Nutr. 2014, 111, 2146–2152. [Google Scholar] [CrossRef]
- Suhr, M.J.; Banjara, N.; Hallen-Adams, H.E. Sequence-Based Methods for Detecting and Evaluating the Human Gut Mycobiome. Lett. Appl. Microbiol. 2016, 62, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Hallen-Adams, H.E.; Kachman, S.D.; Kim, J.; Legge, R.M.; Martínez, I. Fungi Inhabiting the Healthy Human Gastrointestinal Tract: A Diverse and Dynamic Community. Fungal Ecol. 2015, 15, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Pitt, J.I.; Hocking, A.D. Fungi and Food Spoilage, 3rd ed.; Springer: Boston, MA, USA, 2009; ISBN 978-0-387-92206-5. [Google Scholar]
- Widstrom, N.W.; Carr, M.E.; Bagby, M.O.; Black, L.T. Harvest Methods for Estimated Ethanol Yields from Relative Fermentable Carbohydrate Accumulation in Maize Hybrids1. Agron. J. 1987, 79, 758–760. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, L.T.; De Groote, I.; Morales, J.; Barton, N.; Collcutt, S.; Bronk Ramsey, C.; Bouzouggar, A. Earliest Evidence for Caries and Exploitation of Starchy Plant Foods in Pleistocene Hunter-Gatherers from Morocco. Proc. Natl. Acad. Sci. USA 2014, 111, 954–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamad, I.; Sokhna, C.; Raoult, D.; Bittar, F. Molecular Detection of Eukaryotes in a Single Human Stool Sample from Senegal. PLoS ONE 2012, 7, e40888. [Google Scholar] [CrossRef] [Green Version]
- Mar Rodríguez, M.; Pérez, D.; Javier Chaves, F.; Esteve, E.; Marin-Garcia, P.; Xifra, G.; Vendrell, J.; Jové, M.; Pamplona, R.; Ricart, W.; et al. Obesity Changes the Human Gut Mycobiome. Sci. Rep. 2015, 5, 14600. [Google Scholar] [CrossRef]
- Smits, S.A.; Leach, J.; Sonnenburg, E.D.; Gonzalez, C.G.; Lichtman, J.S.; Reid, G.; Knight, R.; Manjurano, A.; Changalucha, J.; Elias, J.E.; et al. Seasonal Cycling in the Gut Microbiome of the Hadza Hunter-Gatherers of Tanzania. Science 2017, 357, 802–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, G.R.; Brown, A.E.; Kiehn, T.E.; Edwards, F.F.; Armstrong, D. Catheter-Related Malassezia Furfur Fungemia in Immunocompromised Patients. Am. J. Med. 1993, 95, 365–370. [Google Scholar] [CrossRef]
- Dupuy, A.K.; David, M.S.; Li, L.; Heider, T.N.; Peterson, J.D.; Montano, E.A.; Dongari-Bagtzoglou, A.; Diaz, P.I.; Strausbaugh, L.D. Redefining the Human Oral Mycobiome with Improved Practices in Amplicon-Based Taxonomy: Discovery of Malassezia as a Prominent Commensal. PLoS ONE 2014, 9, e90899. [Google Scholar] [CrossRef] [Green Version]
- Seddik, H.A.; Ceugniez, A.; Bendali, F.; Cudennec, B.; Drider, D. Yeasts Isolated from Algerian Infants’s Feces Revealed a Burden of Candida Albicans Species, Non-Albicans Candida Species and Saccharomyces Cerevisiae. Arch. Microbiol. 2016, 198, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Findley, K.; Oh, J.; Yang, J.; Conlan, S.; Deming, C.; Meyer, J.A.; Schoenfeld, D.; Nomicos, E.; Park, M.; Kong, H.H.; et al. Topographic Diversity of Fungal and Bacterial Communities in Human Skin. Nature 2013, 498, 367–370. [Google Scholar] [CrossRef]
- Strati, F.; Di Paola, M.; Stefanini, I.; Albanese, D.; Rizzetto, L.; Lionetti, P.; Calabrò, A.; Jousson, O.; Donati, C.; Cavalieri, D.; et al. Age and Gender Affect the Composition of Fungal Population of the Human Gastrointestinal Tract. Front. Microbiol. 2016, 7, 1227. [Google Scholar] [CrossRef]
- Boix Amorós, A.; Puente-Sánchez, F.; du Toit, E.; Linderborg, K.; Zhang, Y.; Yang, B.; Salminen, S.; Isolauri, E.; Tamames, J.; Mira, A.; et al. Mycobiome Profiles in Breast Milk from Healthy Women Depend on Mode of Delivery, Geographic Location, and Interaction with Bacteria. Appl. Environ. Microbiol. 2019, 85. [Google Scholar] [CrossRef] [Green Version]
- Auchtung, T.A.; Fofanova, T.Y.; Stewart, C.J.; Nash, A.K.; Wong, M.C.; Gesell, J.R.; Auchtung, J.M.; Ajami, N.J.; Petrosino, J.F. Investigating Colonization of the Healthy Adult Gastrointestinal Tract by Fungi. mSphere 2018, 3, e00092-18. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, C.; Dollive, S.; Grunberg, S.; Chen, J.; Li, H.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Archaea and Fungi of the Human Gut Microbiome: Correlations with Diet and Bacterial Residents. PLoS ONE 2013, 8, e66019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenman, H.C.; Casadevall, A. Synthesis and Assembly of Fungal Melanin. Appl. Microbiol. Biotechnol. 2012, 93, 931–940. [Google Scholar] [CrossRef] [Green Version]
- Cordero, R.J.; Casadevall, A. Functions of Fungal Melanin beyond Virulence. Fungal Biol. Rev. 2017, 31, 99–112. [Google Scholar] [CrossRef]
- Huet, M.A.L.; Wong, L.W.; Goh, C.; Hussain, M.; Muzahid, N.; Dwiyanto, J.; Lee, S.; Ayub, Q.; Reidpath, D.; Lee, S.M.; et al. Investigation of Culturable Human Gut Mycobiota from the Segamat Community in Johor, Malaysia. World J. Microbiol. Biotechnol. 2021, 37. [Google Scholar] [CrossRef] [PubMed]
- Siegel, P.; Jones, J.G.; Pearsall, D.M.; Wagner, D.P. Environmental and Cultural Correlates in the West Indies: A View from Puerto Rico. In Ancient Borinquen: Archaeology and Ethnohistory of Native Puerto Rico; The University of Alabama Press: Tuscaloosa, AL, USA, 2005; pp. 88–121. [Google Scholar]
- Pagán-Jiménez, J. Las Antillas Precoloniales y Sus Dinámicas Fitoculturales: Evaluando Algunos Viejos Axiomas. Cuba. Arqueológica. Rev. Digit. Arqueol. Cuba. Caribe 2012, 5, 5–19. [Google Scholar]
- Pucu, E.; Russ, J.; Reinhard, K. Diet Analysis Reveals Pre-Historic Meals among the Loma San Gabriel at La Cueva de Los Muertos Chiquitos, Rio Zape, Mexico (600–800 CE). Archaeol. Anthropol. Sci. 2020, 12, 25. [Google Scholar] [CrossRef]
- Jha, A.R.; Davenport, E.R.; Gautam, Y.; Bhandari, D.; Tandukar, S.; Ng, K.M.; Fragiadakis, G.K.; Holmes, S.; Gautam, G.P.; Leach, J.; et al. Gut Microbiome Transition across a Lifestyle Gradient in Himalaya. PLoS Biol. 2018, 16, e2005396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayeni, F.A.; Biagi, E.; Rampelli, S.; Fiori, J.; Soverini, M.; Audu, H.J.; Cristino, S.; Caporali, L.; Schnorr, S.L.; Carelli, V.; et al. Infant and Adult Gut Microbiome and Metabolome in Rural Bassa and Urban Settlers from Nigeria. Cell Rep. 2018, 23, 3056–3067. [Google Scholar] [CrossRef]
- Hallen-Adams, H.E.; Suhr, M.J. Fungi in the Healthy Human Gastrointestinal Tract. Virulence 2017, 8, 352–358. [Google Scholar] [CrossRef]
- Salonen, A.; Salojärvi, J.; Lahti, L.; de Vos, W.M. The Adult Intestinal Core Microbiota Is Determined by Analysis Depth and Health Status. Clin. Microbiol. Infect. 2012, 18, 16–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, Stability and Resilience of the Human Gut Microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Suhr, M.J.; Hallen-Adams, H.E. The Human Gut Mycobiome: Pitfalls and Potentials—A Mycologist’s Perspective. Mycologia 2015, 107, 1057–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angebault, C.; Djossou, F.; Abélanet, S.; Permal, E.; Diancourt, L.; Bouchier, C.; Woerther, P.-L.; Catzeflis, F.; Andremont, A.; d’Enfert, C.; et al. Candida Albicans Is Not Always the Preferential Yeast Colonizing Humans: A Study in Wayampi Amerindians. J. Infect. Dis. 2013, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| diff.btw | diff.win | Effect | wi.ep | wi.eBH | Genus | |
|---|---|---|---|---|---|---|
| 1 | −6.68239 | 2.648387 | −2.44229 | 1.11 × 10−5 | 0.005524 | Aspergillus |
| 2 | −4.12151 | 2.869157 | −1.4792 | 2.81 × 10−5 | 0.00606 | Penicillium |
| 3 | −4.1763 | 2.971508 | −1.4539 | 3.07 × 10−5 | 0.006401 | Rasamsonia |
| 4 | −4.44338 | 3.153649 | −1.4055 | 3.57 × 10−5 | 0.006584 | Byssochlamys |
| 5 | −2.98222 | 2.36935 | −1.26018 | 0.000116 | 0.013226 | Talaromyces |
| 6 | −3.63104 | 3.269716 | −1.14088 | 0.000141 | 0.014606 | Blastomyces |
| 7 | −3.90086 | 3.668713 | −1.13541 | 0.000667 | 0.037039 | Monascus |
| 8 | −4.02314 | 3.927779 | −1.02516 | 0.000609 | 0.033089 | Penicilliopsis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reynoso-García, J.; Narganes-Storde, Y.; Santiago-Rodriguez, T.M.; Toranzos, G.A. Mycobiome-Host Coevolution? The Mycobiome of Ancestral Human Populations Seems to Be Different and Less Diverse Than Those of Extant Native and Urban-Industrialized Populations. Microorganisms 2022, 10, 459. https://doi.org/10.3390/microorganisms10020459
Reynoso-García J, Narganes-Storde Y, Santiago-Rodriguez TM, Toranzos GA. Mycobiome-Host Coevolution? The Mycobiome of Ancestral Human Populations Seems to Be Different and Less Diverse Than Those of Extant Native and Urban-Industrialized Populations. Microorganisms. 2022; 10(2):459. https://doi.org/10.3390/microorganisms10020459
Chicago/Turabian StyleReynoso-García, Jelissa, Yvonne Narganes-Storde, Tasha M. Santiago-Rodriguez, and Gary A. Toranzos. 2022. "Mycobiome-Host Coevolution? The Mycobiome of Ancestral Human Populations Seems to Be Different and Less Diverse Than Those of Extant Native and Urban-Industrialized Populations" Microorganisms 10, no. 2: 459. https://doi.org/10.3390/microorganisms10020459
APA StyleReynoso-García, J., Narganes-Storde, Y., Santiago-Rodriguez, T. M., & Toranzos, G. A. (2022). Mycobiome-Host Coevolution? The Mycobiome of Ancestral Human Populations Seems to Be Different and Less Diverse Than Those of Extant Native and Urban-Industrialized Populations. Microorganisms, 10(2), 459. https://doi.org/10.3390/microorganisms10020459