
Is There a Role for Gut Microbiome Dysbiosis in IgA Nephropathy?


{kind=link}
Abstract
:1. The IgA System and Mucosal Immunity
2. Pathophysiology of IgA Nephropathy
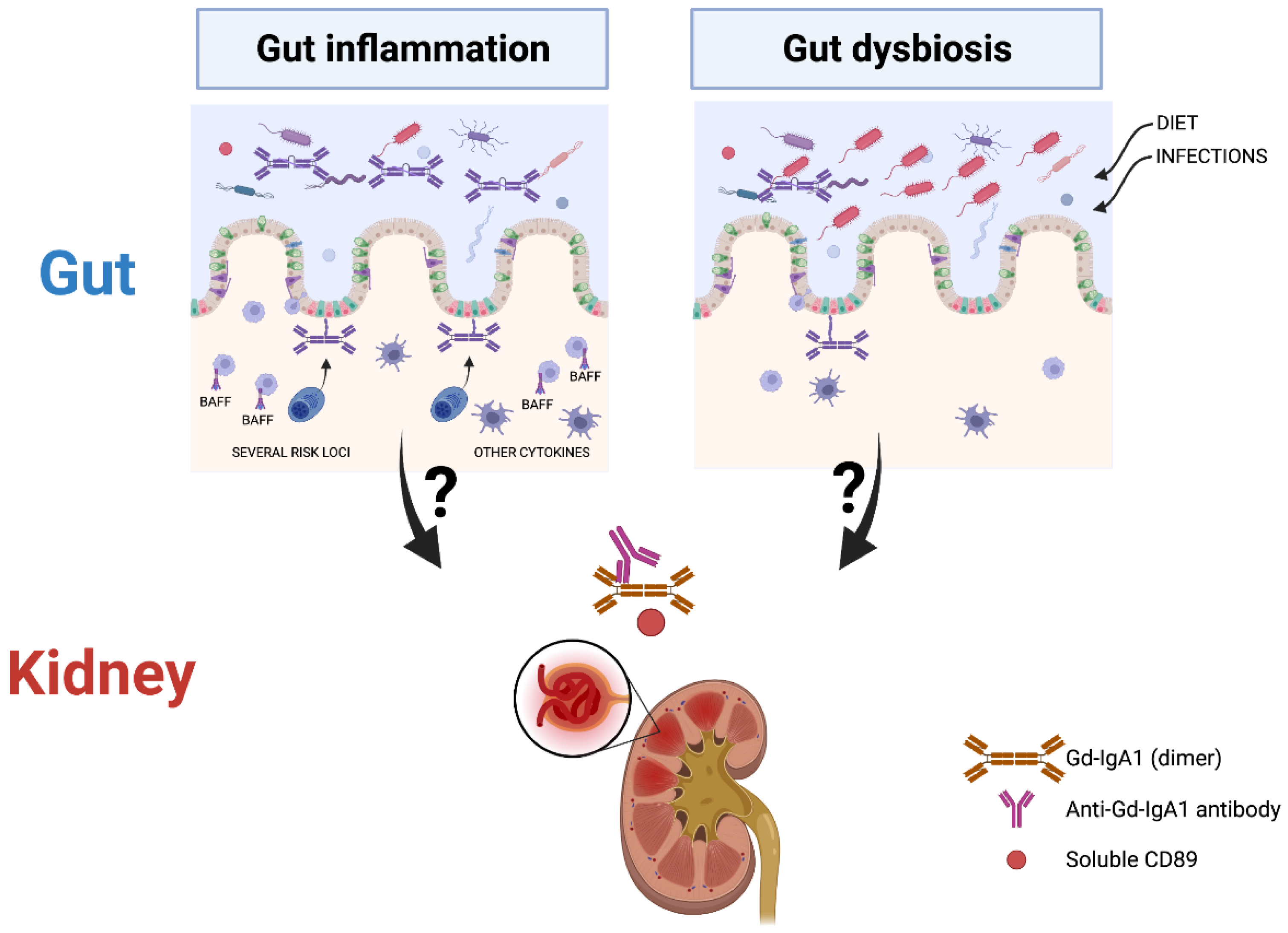
3. Evidence for the Role of a Gut-Kidney Inflammatory Axis in IgAN
4. Role of Food Antigens and Infections in IgAN
5. Which Role for Gut Microbial Dysbiosis in IgAN?
6. Therapeutic Approaches Targeting the Microbiota
7. Conclusions and Hypotheses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gutzeit, C.; Magri, G.; Cerutti, A. Intestinal IgA production and its role in host-microbe interaction. Immunol. Rev. 2014, 260, 76–85. [Google Scholar] [CrossRef]
- Kerr, M.A. The structure and function of human IgA. Biochem. J. 1990, 271, 285–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, R.C.; Van De Winkel, J.G. IgA Fc receptors. Annu. Rev. Immunol. 2003, 21, 177–204. [Google Scholar] [CrossRef] [PubMed]
- Kaetzel, C.S.; Mestecky, J.; Johansen, F.E. Two Cells, One Antibody: The Discovery of the Cellular Origins and Transport of Secretory IgA. J. Immunol. 2017, 198, 1765–1767. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.W.; Sibley, D.A.; Nikolova, E.B.; Tomana, M.; Mestecky, J. IgA antibody as a non-inflammatory regulator of immunity. Biochem. Soc. Trans. 1997, 25, 466–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquier, B.; Launay, P.; Kanamaru, Y.; Moura, I.C.; Pfirsch, S.; Ruffie, C.; Henin, D.; Benhamou, M.; Pretolani, M.; Blank, U.; et al. Identification of FcalphaRI as an inhibitory receptor that controls inflammation: Dual role of FcRgamma ITAM. Immunity 2005, 22, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossato, E.; Ben Mkaddem, S.; Kanamaru, Y.; Hurtado-Nedelec, M.; Hayem, G.; Descatoire, V.; Vonarburg, C.; Miescher, S.; Zuercher, A.W.; Monteiro, R.C. Reversal of Arthritis by Human Monomeric IgA Through the Receptor-Mediated SH2 Domain-Containing Phosphatase 1 Inhibitory Pathway. Arthritis Rheumatol. 2015, 67, 1766–1777. [Google Scholar] [CrossRef] [Green Version]
- Jacob, C.M.; Pastorino, A.C.; Fahl, K.; Carneiro-Sampaio, M.; Monteiro, R.C. Autoimmunity in IgA deficiency: Revisiting the role of IgA as a silent housekeeper. J. Clin. Immunol. 2008, 28 (Suppl. S1), S56–S61. [Google Scholar] [CrossRef]
- Monteiro, R.C. Role of IgA and IgA fc receptors in inflammation. J. Clin. Immunol. 2010, 30, 1–9. [Google Scholar] [CrossRef]
- Bunker, J.J.; Bendelac, A. IgA Responses to Microbiota. Immunity 2018, 49, 211–224. [Google Scholar] [CrossRef] [Green Version]
- Kiyono, H.; Fukuyama, S. NALT- versus Peyer’s-patch-mediated mucosal immunity. Nat. Rev. Immunol. 2004, 4, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Rochereau, N.; Drocourt, D.; Perouzel, E.; Pavot, V.; Redelinghuys, P.; Brown, G.D.; Tiraby, G.; Roblin, X.; Verrier, B.; Genin, C.; et al. Dectin-1 is essential for reverse transcytosis of glycosylated SIgA-antigen complexes by intestinal M cells. PLoS Biol. 2013, 11, e1001658. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Magri, G.; Grasset, E.K.; Cerutti, A. Rethinking mucosal antibody responses: IgM, IgG and IgD join IgA. Nat. Rev. Immunol. 2020, 20, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Fagarasan, S.; Kawamoto, S.; Kanagawa, O.; Suzuki, K. Adaptive immune regulation in the gut: T cell-dependent and T cell-independent IgA synthesis. Annu. Rev. Immunol. 2010, 28, 243–273. [Google Scholar] [CrossRef]
- Grasset, E.K.; Chorny, A.; Casas-Recasens, S.; Gutzeit, C.; Bongers, G.; Thomsen, I.; Chen, L.; He, Z.; Matthews, D.B.; Oropallo, M.A.; et al. Gut T cell-independent IgA responses to commensal bacteria require engagement of the TACI receptor on B cells. Sci. Immunol. 2020, 5, eaat7117. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Motta, J.P.; Wallace, J.L.; Buret, A.G.; Deraison, C.; Vergnolle, N. Gastrointestinal biofilms in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 314–334. [Google Scholar] [CrossRef]
- Brandl, C.; Bucci, L.; Schett, G.; Zaiss, M.M. Crossing the barriers: Revisiting the gut feeling in rheumatoid arthritis. Eur. J. Immunol. 2021, 51, 798–810. [Google Scholar] [CrossRef]
- Weis, A.M.; Round, J.L. Microbiota-antibody interactions that regulate gut homeostasis. Cell Host Microbe 2021, 29, 334–346. [Google Scholar] [CrossRef]
- Bunker, J.J.; Erickson, S.A.; Flynn, T.M.; Henry, C.; Koval, J.C.; Meisel, M.; Jabri, B.; Antonopoulos, D.A.; Wilson, P.C.; Bendelac, A. Natural polyreactive IgA antibodies coat the intestinal microbiota. Science 2017, 358, eaan6619. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, D.B.; Suzuki, K.; Fagarasan, S. Fostering of advanced mutualism with gut microbiota by Immunoglobulin A. Immunol. Rev. 2016, 270, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Hinglais, N. Intercapillary deposits of IgA-IgG. J. Urol. Nephrol. 1968, 74, 694–695. [Google Scholar]
- Monteiro, R.C. Recent advances in the physiopathology of IgA nephropathy. Nephrol. Ther. 2018, 14 (Suppl. S1), S1–S8. [Google Scholar] [CrossRef] [PubMed]
- Rifai, A.; Small, P.A., Jr.; Teague, P.O.; Ayoub, E.M. Experimental IgA nephropathy. J. Exp. Med. 1979, 150, 1161–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.; Wong, S.S.; Rifai, A. Glomerular immune deposits in experimental IgA nephropathy. A continuum of circulating and in situ formed immune complexes. Am. J. Pathol. 1988, 130, 216–222. [Google Scholar]
- Isaacs, K.L.; Miller, F. Role of antigen size and charge in immune complex glomerulonephritis. Lab. Investig. 1982, 47, 198–205. [Google Scholar]
- Tomino, Y.; Sakai, H.; Miura, M.; Endoh, M.; Nomoto, Y. Detection of polymeric IgA in glomeruli from patients with IgA nephropathy. Clin. Exp. Immunol. 1982, 49, 419–425. [Google Scholar]
- Monteiro, R.C.; Halbwachs-Mecarelli, L.; Roque-Barreira, M.C.; Noel, L.H.; Berger, J.; Lesavre, P. Charge and size of mesangial IgA in IgA nephropathy. Kidney Int. 1985, 28, 666–671. [Google Scholar] [CrossRef] [Green Version]
- Mestecky, J.; Raska, M.; Julian, B.A.; Gharavi, A.G.; Renfrow, M.B.; Moldoveanu, Z.; Novak, L.; Matousovic, K.; Novak, J. IgA nephropathy: Molecular mechanisms of the disease. Annu. Rev. Pathol. 2013, 8, 217–240. [Google Scholar] [CrossRef] [Green Version]
- Allen, A.C.; Bailey, E.M.; Brenchley, P.E.; Buck, K.S.; Barratt, J.; Feehally, J. Mesangial IgA1 in IgA nephropathy exhibits aberrant O-glycosylation: Observations in three patients. Kidney Int. 2001, 60, 969–973. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Fan, R.; Zhang, Z.; Brown, R.; Hall, S.; Julian, B.A.; Chatham, W.W.; Suzuki, Y.; Wyatt, R.J.; Moldoveanu, Z.; et al. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J. Clin. Investig. 2009, 119, 1668–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizk, D.V.; Saha, M.K.; Hall, S.; Novak, L.; Brown, R.; Huang, Z.Q.; Fatima, H.; Julian, B.A.; Novak, J. Glomerular Immunodeposits of Patients with IgA Nephropathy Are Enriched for IgG Autoantibodies Specific for Galactose-Deficient IgA1. J. Am. Soc. Nephrol. 2019, 30, 2017–2026. [Google Scholar] [CrossRef] [PubMed]
- Kiryluk, K.; Li, Y.; Moldoveanu, Z.; Suzuki, H.; Reily, C.; Hou, P.; Xie, J.; Mladkova, N.; Prakash, S.; Fischman, C.; et al. GWAS for serum galactose-deficient IgA1 implicates critical genes of the O-glycosylation pathway. PLoS Genet. 2017, 13, e1006609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gale, D.P.; Molyneux, K.; Wimbury, D.; Higgins, P.; Levine, A.P.; Caplin, B.; Ferlin, A.; Yin, P.; Nelson, C.P.; Stanescu, H.; et al. Galactosylation of IgA1 Is Associated with Common Variation in C1GALT1. J. Am. Soc. Nephrol. 2017, 28, 2158–2166. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.N.; Zhou, X.J.; Chen, P.; Yu, G.Z.; Zhang, X.; Hou, P.; Liu, L.J.; Shi, S.F.; Lv, J.C.; Zhang, H. Interaction between G ALNT12 and C1GALT1 Associates with Galactose-Deficient IgA1 and IgA Nephropathy. J. Am. Soc. Nephrol. 2021, 32, 545–552. [Google Scholar] [CrossRef]
- Moura, I.C.; Arcos-Fajardo, M.; Sadaka, C.; Leroy, V.; Benhamou, M.; Novak, J.; Vrtovsnik, F.; Haddad, E.; Chintalacharuvu, K.R.; Monteiro, R.C. Glycosylation and size of IgA1 are essential for interaction with mesangial transferrin receptor in IgA nephropathy. J. Am. Soc. Nephrol. 2004, 15, 622–634. [Google Scholar] [CrossRef] [Green Version]
- Kashem, A.; Endoh, M.; Yano, N.; Yamauchi, F.; Nomoto, Y.; Sakai, H.; Kurokawa, K. Glomerular Fc alphaR expression and disease activity in IgA nephropathy. Am. J. Kidney Dis. 1997, 30, 389–396. [Google Scholar] [CrossRef]
- Kanamaru, Y.; Arcos-Fajardo, M.; Moura, I.C.; Tsuge, T.; Cohen, H.; Essig, M.; Vrtovsnik, F.; Loirat, C.; Peuchmaur, M.; Beaudoin, L.; et al. Fc alpha receptor I activation induces leukocyte recruitment and promotes aggravation of glomerulonephritis through the FcR gamma adaptor. Eur. J. Immunol. 2007, 37, 1116–1128. [Google Scholar] [CrossRef]
- Grossetete, B.; Launay, P.; Lehuen, A.; Jungers, P.; Bach, J.F.; Monteiro, R.C. Down-regulation of Fc alpha receptors on blood cells of IgA nephropathy patients: Evidence for a negative regulatory role of serum IgA. Kidney Int. 1998, 53, 1321–1335. [Google Scholar] [CrossRef] [Green Version]
- Launay, P.; Grossetete, B.; Arcos-Fajardo, M.; Gaudin, E.; Torres, S.P.; Beaudoin, L.; Patey-Mariaud de Serre, N.; Lehuen, A.; Monteiro, R.C. Fcalpha receptor (CD89) mediates the development of immunoglobulin A (IgA) nephropathy (Berger’s disease). Evidence for pathogenic soluble receptor-Iga complexes in patients and CD89 transgenic mice. J. Exp. Med. 2000, 191, 1999–2009. [Google Scholar] [CrossRef] [Green Version]
- Vuong, M.T.; Hahn-Zoric, M.; Lundberg, S.; Gunnarsson, I.; van Kooten, C.; Wramner, L.; Seddighzadeh, M.; Fernstrom, A.; Hanson, L.A.; Do, L.T.; et al. Association of soluble CD89 levels with disease progression but not susceptibility in IgA nephropathy. Kidney Int. 2010, 78, 1281–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthelot, L.; Papista, C.; Maciel, T.T.; Biarnes-Pelicot, M.; Tissandie, E.; Wang, P.H.; Tamouza, H.; Jamin, A.; Bex-Coudrat, J.; Gestin, A.; et al. Transglutaminase is essential for IgA nephropathy development acting through IgA receptors. J. Exp. Med. 2012, 209, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Berthelot, L.; Robert, T.; Vuiblet, V.; Tabary, T.; Braconnier, A.; Drame, M.; Toupance, O.; Rieu, P.; Monteiro, R.C.; Toure, F. Recurrent IgA nephropathy is predicted by altered glycosylated IgA, autoantibodies and soluble CD89 complexes. Kidney Int. 2015, 88, 815–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cambier, A.; Gleeson, P.J.; Abbad, L.; Canesi, F.; da Silva, J.; Bex-Coudrat, J.; Deschenes, G.; Boyer, O.; Rabant, M.; Ulinski, T.; et al. Soluble CD89 is a critical factor for mesangial proliferation in childhood IgA nephropathy. Kidney Int. 2021, 101, 274–287. [Google Scholar] [CrossRef] [PubMed]
- Floege, J.; Feehally, J. The mucosa-kidney axis in IgA nephropathy. Nat. Rev. Nephrol. 2016, 12, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Kiryluk, K.; Li, Y.; Scolari, F.; Sanna-Cherchi, S.; Choi, M.; Verbitsky, M.; Fasel, D.; Lata, S.; Prakash, S.; Shapiro, S.; et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat. Genet. 2014, 46, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Davin, J.C.; Forget, P.; Mahieu, P.R. Increased intestinal permeability to (51 Cr) EDTA is correlated with IgA immune complex-plasma levels in children with IgA-associated nephropathies. Acta Paediatr. Scand. 1988, 77, 118–124. [Google Scholar] [CrossRef]
- Rostoker, G.; Wirquin, V.; Terzidis, H.; Petit-Phar, M.; Chaumette, M.T.; Delchier, J.C.; Belghiti, D.; Lang, P.; Dubert, J.M.; Meignan, M.; et al. Mucosal immunity in primary glomerulonephritis. III. Study of intestinal permeability. Nephron 1993, 63, 286–290. [Google Scholar] [CrossRef]
- Kovacs, T.; Kun, L.; Schmelczer, M.; Wagner, L.; Davin, J.C.; Nagy, J. Do intestinal hyperpermeability and the related food antigens play a role in the progression of IgA nephropathy? I. Study of intestinal permeability. Am. J. Nephrol. 1996, 16, 500–505. [Google Scholar] [CrossRef]
- Rehnberg, J.; Symreng, A.; Ludvigsson, J.F.; Emilsson, L. Inflammatory Bowel Disease Is More Common in Patients with IgA Nephropathy and Predicts Progression of ESKD: A Swedish Population-Based Cohort Study. J. Am. Soc. Nephrol. 2021, 32, 411–423. [Google Scholar] [CrossRef]
- Fellstrom, B.C.; Barratt, J.; Cook, H.; Coppo, R.; Feehally, J.; de Fijter, J.W.; Floege, J.; Hetzel, G.; Jardine, A.G.; Locatelli, F.; et al. Targeted-release budesonide versus placebo in patients with IgA nephropathy (NEFIGAN): A double-blind, randomised, placebo-controlled phase 2b trial. Lancet 2017, 389, 2117–2127. [Google Scholar] [CrossRef] [Green Version]
- Emancipator, S.N.; Gallo, G.R.; Lamm, M.E. Experimental IgA nephropathy induced by oral immunization. J. Exp. Med. 1983, 157, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Sieniawska, M.; Beutner, E.H.; Chorzelski, T.P. Are immunological markers of gluten-sensitive enteropathy detectable in IgA nephropathy? Lancet 1988, 2, 1307. [Google Scholar] [CrossRef]
- Smerud, H.K.; Fellstrom, B.; Hallgren, R.; Osagie, S.; Venge, P.; Kristjansson, G. Gluten sensitivity in patients with IgA nephropathy. Nephrol. Dial. Transplant. 2009, 24, 2476–2481. [Google Scholar] [CrossRef] [Green Version]
- Papista, C.; Lechner, S.; Ben Mkaddem, S.; LeStang, M.B.; Abbad, L.; Bex-Coudrat, J.; Pillebout, E.; Chemouny, J.M.; Jablonski, M.; Flamant, M.; et al. Gluten exacerbates IgA nephropathy in humanized mice through gliadin-CD89 interaction. Kidney Int. 2015, 88, 276–285. [Google Scholar] [CrossRef]
- Coppo, R.; Amore, A.; Roccatello, D. Dietary antigens and primary immunoglobulin A nephropathy. J. Am. Soc. Nephrol. 1992, 2, S173–S180. [Google Scholar] [CrossRef]
- Kloster Smerud, H.; Fellstrom, B.; Hallgren, R.; Osagie, S.; Venge, P.; Kristjansson, G. Gastrointestinal sensitivity to soy and milk proteins in patients with IgA nephropathy. Clin. Nephrol. 2010, 74, 364–371. [Google Scholar] [CrossRef]
- Hinoshita, F.; Suzuki, Y.; Yokoyama, K.; Hara, S.; Yamada, A.; Ogura, Y.; Hashimoto, H.; Tomura, S.; Marumo, F.; Ueno, Y. Experimental IgA nephropathy induced by a low-dose environmental mycotoxin, nivalenol. Nephron 1997, 75, 469–478. [Google Scholar] [CrossRef]
- Sevillano, A.M.; Gutierrez, E.; Yuste, C.; Cavero, T.; Merida, E.; Rodriguez, P.; Garcia, A.; Morales, E.; Fernandez, C.; Martinez, M.A.; et al. Remission of Hematuria Improves Renal Survival in IgA Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 3089–3099. [Google Scholar] [CrossRef]
- Koyama, A.; Kobayashi, M.; Yamaguchi, N.; Yamagata, K.; Takano, K.; Nakajima, M.; Irie, F.; Goto, M.; Igarashi, M.; Iitsuka, T.; et al. Glomerulonephritis associated with MRSA infection: A possible role of bacterial superantigen. Kidney Int. 1995, 47, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Satoskar, A.A.; Nadasdy, G.; Plaza, J.A.; Sedmak, D.; Shidham, G.; Hebert, L.; Nadasdy, T. Staphylococcus infection-associated glomerulonephritis mimicking IgA nephropathy. Clin. J. Am. Soc. Nephrol. 2006, 1, 1179–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharmin, S.; Shimizu, Y.; Hagiwara, M.; Hirayama, K.; Koyama, A. Staphylococcus aureus antigens induce IgA-type glomerulonephritis in Balb/c mice. J. Nephrol. 2004, 17, 504–511. [Google Scholar] [PubMed]
- Suzuki, S.; Nakatomi, Y.; Sato, H.; Tsukada, H.; Arakawa, M. Haemophilus parainfluenzae antigen and antibody in renal biopsy samples and serum of patients with IgA nephropathy. Lancet 1994, 343, 12–16. [Google Scholar] [CrossRef]
- Yamamoto, C.; Suzuki, S.; Kimura, H.; Yoshida, H.; Gejyo, F. Experimental nephropathy induced by Haemophilus parainfluenzae antigens. Nephron 2002, 90, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Portis, J.L.; Coe, J.E. Deposition of IgA in renal glomeruli of mink affected with Aleutian disease. Am. J. Pathol. 1979, 96, 227–236. [Google Scholar]
- Jessen, R.H.; Emancipator, S.N.; Jacobs, G.H.; Nedrud, J.G. Experimental IgA-IgG nephropathy induced by a viral respiratory pathogen. Dependence on antigen form and immune status. Lab. Investig. 1992, 67, 379–386. [Google Scholar]
- Suzuki, H.; Suzuki, Y.; Narita, I.; Aizawa, M.; Kihara, M.; Yamanaka, T.; Kanou, T.; Tsukaguchi, H.; Novak, J.; Horikoshi, S.; et al. Toll-like receptor 9 affects severity of IgA nephropathy. J. Am. Soc. Nephrol. 2008, 19, 2384–2395. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, D.D.; Kujawa, J.; Wilson, C.; Papandile, A.; Poreci, U.; Porfilio, E.A.; Ward, L.; Lawson, M.A.; Macpherson, A.J.; McCoy, K.D.; et al. Mice overexpressing BAFF develop a commensal flora-dependent, IgA-associated nephropathy. J. Clin. Investig. 2011, 121, 3991–4002. [Google Scholar] [CrossRef] [Green Version]
- De Angelis, M.; Montemurno, E.; Piccolo, M.; Vannini, L.; Lauriero, G.; Maranzano, V.; Gozzi, G.; Serrazanetti, D.; Dalfino, G.; Gobbetti, M.; et al. Microbiota and metabolome associated with immunoglobulin A nephropathy (IgAN). PLoS ONE 2014, 9, e99006. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Dong, R.; Bai, M.; Zhao, J.; Wang, D.; Ning, X.; Sun, S. A Comparative Study of the Gut Microbiota Associated With Immunoglobulin a Nephropathy and Membranous Nephropathy. Front. Cell. Infect. Microbiol. 2020, 10, 557368. [Google Scholar] [CrossRef] [PubMed]
- Sallustio, F.; Curci, C.; Chaoul, N.; Fonto, G.; Lauriero, G.; Picerno, A.; Divella, C.; Di Leo, V.; De Angelis, M.; Ben Mkaddem, S.; et al. High levels of gut-homing immunoglobulin A + B lymphocytes support the pathogenic role of intestinal mucosal hyperresponsiveness in immunoglobulin A nephropathy patients. Nephrol. Dial. Transplant. 2021, 36, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Gesualdo, L.; Di Leo, V.; Coppo, R. The mucosal immune system and IgA nephropathy. Semin. Immunopathol. 2021, 43, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Coppo, R.; Camilla, R.; Amore, A.; Peruzzi, L.; Dapra, V.; Loiacono, E.; Vatrano, S.; Rollino, C.; Sepe, V.; Rampino, T.; et al. Toll-like receptor 4 expression is increased in circulating mononuclear cells of patients with immunoglobulin A nephropathy. Clin. Exp. Immunol. 2010, 159, 73–81. [Google Scholar] [CrossRef]
- Yoon, H.J.; Shin, J.H.; Yang, S.H.; Chae, D.W.; Kim, H.; Lee, D.S.; Kim, H.L.; Kim, S.; Lee, J.S.; Kim, Y.S. Association of the CD14 gene -159C polymorphism with progression of IgA nephropathy. J. Med. Genet. 2003, 40, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Currie, E.G.; Coburn, B.; Porfilio, E.A.; Lam, P.; Rojas, O.L.; Novak, J.; Yang, S.; Chowdhury, R.B.; Ward, L.A.; Wang, P.W.; et al. Immunoglobulin A nephropathy is characterized by anti-commensal humoral immune responses. JCI Insight 2022, 7, e141289. [Google Scholar] [CrossRef]
- Monteiro, R.C.; Suzuki, Y. Are there animal models of IgA nephropathy? Semin. Immunopathol. 2021, 43, 639–648. [Google Scholar] [CrossRef]
- Oruc, Z.; Oblet, C.; Boumediene, A.; Druilhe, A.; Pascal, V.; Le Rumeur, E.; Cuvillier, A.; El Hamel, C.; Lecardeur, S.; Leanderson, T.; et al. IgA Structure Variations Associate with Immune Stimulations and IgA Mesangial Deposition. J. Am. Soc. Nephrol. 2016, 27, 2748–2761. [Google Scholar] [CrossRef] [Green Version]
- Chemouny, J.M.; Gleeson, P.J.; Abbad, L.; Lauriero, G.; Boedec, E.; Le Roux, K.; Monot, C.; Bredel, M.; Bex-Coudrat, J.; Sannier, A.; et al. Modulation of the microbiota by oral antibiotics treats immunoglobulin A nephropathy in humanized mice. Nephrol. Dial. Transplant. 2019, 34, 1135–1144. [Google Scholar] [CrossRef]
- Di Leo, V.; Gleeson, P.J.; Sallustio, F.; Bounaix, C.; Da Silva, J.; Loreto, G.; Ben Mkaddem, S.; Monteiro, R.C. Rifaximin as a Potential Treatment for IgA Nephropathy in a Humanized Mice Model. J. Pers. Med. 2021, 11, 309. [Google Scholar] [CrossRef]
- Vitetta, L.; Vitetta, G.; Hall, S. Immunological Tolerance and Function: Associations Between Intestinal Bacteria, Probiotics, Prebiotics, and Phages. Front. Immunol. 2018, 9, 2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guida, B.; Germano, R.; Trio, R.; Russo, D.; Memoli, B.; Grumetto, L.; Barbato, F.; Cataldi, M. Effect of short-term synbiotic treatment on plasma p-cresol levels in patients with chronic renal failure: A randomized clinical trial. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Soylu, A.; Berktas, S.; Sarioglu, S.; Erbil, G.; Yilmaz, O.; Demir, B.K.; Tufan, Y.; Yesilirmak, D.; Turkmen, M.; Kavukcu, S. Saccharomyces boulardii prevents oral-poliovirus vaccine-induced IgA nephropathy in mice. Pediatr. Nephrol. 2008, 23, 1287–1291. [Google Scholar] [CrossRef] [PubMed]
- Staley, C.; Hamilton, M.J.; Vaughn, B.P.; Graiziger, C.T.; Newman, K.M.; Kabage, A.J.; Sadowsky, M.J.; Khoruts, A. Successful Resolution of Recurrent Clostridium difficile Infection using Freeze-Dried, Encapsulated Fecal Microbiota; Pragmatic Cohort Study. Am. J. Gastroenterol. 2017, 112, 940–947. [Google Scholar] [CrossRef] [Green Version]
- Lauriero, G.; Abbad, L.; Vacca, M.; Celano, G.; Chemouny, J.M.; Calasso, M.; Berthelot, L.; Gesualdo, L.; De Angelis, M.; Monteiro, R.C. Fecal Microbiota Transplantation Modulates Renal Phenotype in the Humanized Mouse Model of IgA Nephropathy. Front. Immunol. 2021, 12, 694787. [Google Scholar] [CrossRef]
- Barba, C.; Soulage, C.O.; Caggiano, G.; Glorieux, G.; Fouque, D.; Koppe, L. Effects of Fecal Microbiota Transplantation on Composition in Mice with CKD. Toxins 2020, 12, 741. [Google Scholar] [CrossRef]
- Du, L.; Ma, L.; Qi, F.; Zheng, X.; Jiang, C.; Li, A.; Wan, X.; Liu, S.J.; Li, S. Characterization of a Unique Pathway for 4-Cresol Catabolism Initiated by Phosphorylation in Corynebacterium glutamicum. J. Biol. Chem. 2016, 291, 6583–6594. [Google Scholar] [CrossRef] [Green Version]
- Qin, W.; Zhong, X.; Fan, J.M.; Zhang, Y.J.; Liu, X.R.; Ma, X.Y. External suppression causes the low expression of the Cosmc gene in IgA nephropathy. Nephrol. Dial. Transplant. 2008, 23, 1608–1614. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Raska, M.; Yamada, K.; Moldoveanu, Z.; Julian, B.A.; Wyatt, R.J.; Tomino, Y.; Gharavi, A.G.; Novak, J. Cytokines alter IgA1 O-glycosylation by dysregulating C1GalT1 and ST6GalNAc-II enzymes. J. Biol. Chem. 2014, 289, 5330–5339. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monteiro, R.C.; Rafeh, D.; Gleeson, P.J. Is There a Role for Gut Microbiome Dysbiosis in IgA Nephropathy? Microorganisms 2022, 10, 683. https://doi.org/10.3390/microorganisms10040683
Monteiro RC, Rafeh D, Gleeson PJ. Is There a Role for Gut Microbiome Dysbiosis in IgA Nephropathy? Microorganisms. 2022; 10(4):683. https://doi.org/10.3390/microorganisms10040683
Chicago/Turabian StyleMonteiro, Renato C., Dina Rafeh, and Patrick J. Gleeson. 2022. "Is There a Role for Gut Microbiome Dysbiosis in IgA Nephropathy?" Microorganisms 10, no. 4: 683. https://doi.org/10.3390/microorganisms10040683
APA StyleMonteiro, R. C., Rafeh, D., & Gleeson, P. J. (2022). Is There a Role for Gut Microbiome Dysbiosis in IgA Nephropathy? Microorganisms, 10(4), 683. https://doi.org/10.3390/microorganisms10040683