
IntegronFinder 2.0: Identification and Analysis of Integrons across Bacteria, with a Focus on Antibiotic Resistance in Klebsiella

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Refactoring of IntegronFinder
2.2. Novel Functionalities
2.3. Input
2.4. Output
2.5. Availability
2.6. RefSeq Complete Genomes
2.7. Klebsiella Data
2.8. Analysis of Antibiotic Resistance Genes (ARGs)
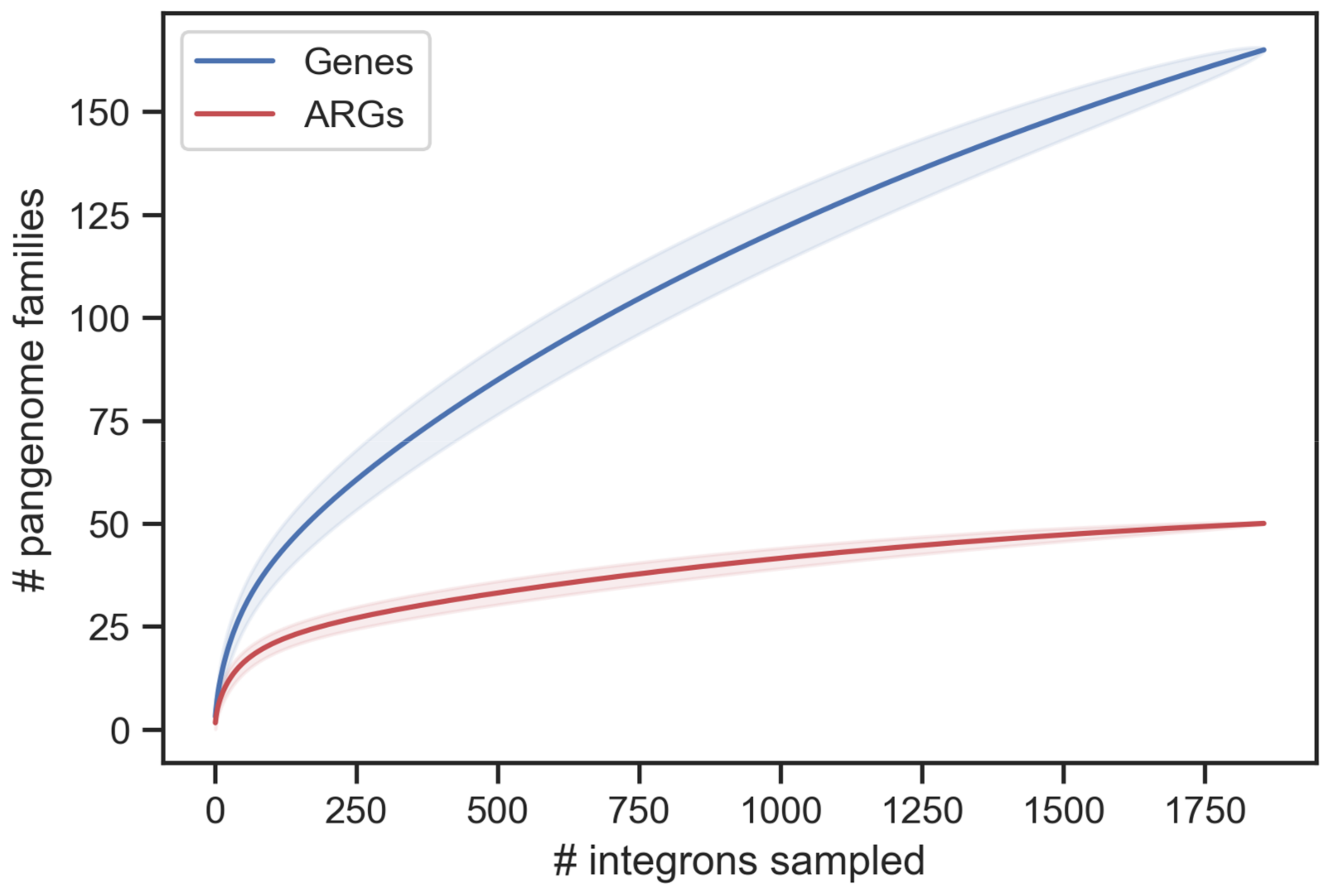
2.9. Saturation Curves
2.10. Graphics and Visualizations
3. Results and Discussion
3.1. Distribution of Integrons across Bacteria
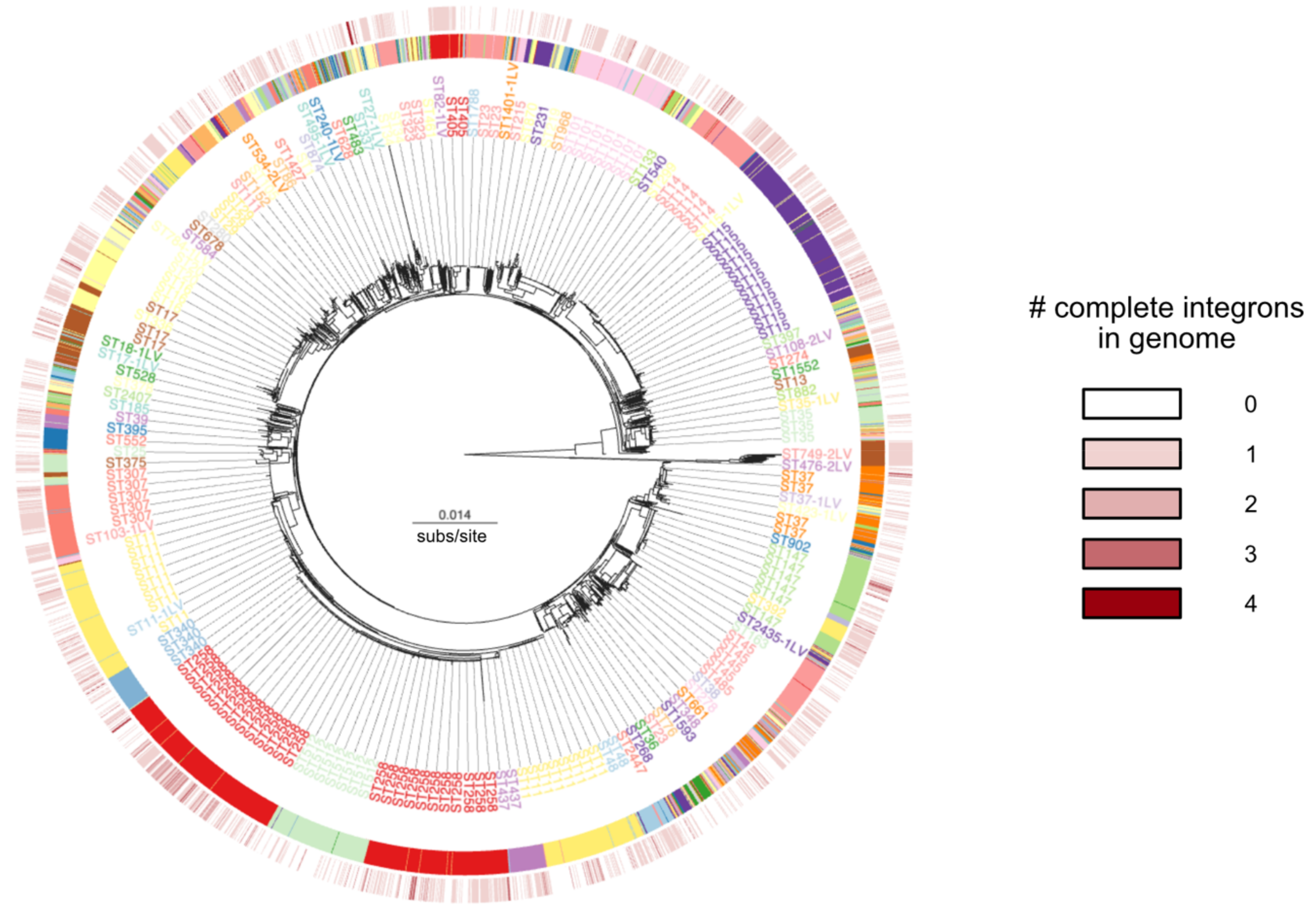
3.2. Recent Spread of Integrons in K. pneumoniae
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haudiquet, M.; de Sousa, J.M.; Touchon, M.; Rocha, E. Selfish, promiscuous, and sometimes useful: How mobile genetic elements drive horizontal gene transfer in microbial populations. EcoEvoRxiv 2021. preprint. [Google Scholar] [CrossRef]
- Arnold, B.J.; Huang, I.-T.; Hanage, W.P. Horizontal gene transfer and adaptive evolution in bacteria. Nat. Rev. Genet. 2021, 20, 206–218. [Google Scholar] [CrossRef]
- Cerveau, N.; Leclercq, S.; Bouchon, D.; Cordaux, R. Evolutionary Dynamics and Genomic Impact of Prokaryote Transposable Elements. In Evolutionary Biology—Concepts, Biodiversity, Macroevolution and Genome Evolution; Pontarotti, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 291–312. [Google Scholar]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef] [PubMed]
- Cambray, G.; Guerout, A.M.; Mazel, D. Integrons. Annu. Rev. Genet. 2010, 44, 141–166. [Google Scholar]
- Escudero, J.A.; Loot, C.; Nivina, A.; Mazel, D. The Integron: Adaptation on Demand. Microbiol. Spectr. 2015, 3, 139–161. [Google Scholar] [CrossRef]
- Bouvier, M.; Demarre, G.; Mazel, D. Integron cassette insertion: A recombination process involving a folded single strand substrate. EMBO J. 2005, 24, 4356–4367. [Google Scholar] [CrossRef]
- Smyshlyaev, G.; Bateman, A.; Barabas, O. Sequence analysis of tyrosine recombinases allows annotation of mobile genetic elements in prokaryotic genomes. Mol. Syst. Biol. 2021, 17, e9880. [Google Scholar] [CrossRef]
- Nivina, A.; Grieb, M.S.; Loot, C.; Bikard, D.; Cury, J.; Shehata, L.; Bernardes, J.; Mazel, D. Structure-specific DNA recombination sites: Design, validation, and machine learning–based refinement. Sci. Adv. 2020, 6, eaay2922. [Google Scholar] [CrossRef]
- Cury, J.; Jové, T.; Touchon, M.; Néron, B.; Rocha, E. Identification and analysis of integrons and cassette arrays in bacterial genomes. Nucl. Acids Res. 2016, 44, 4539–4550. [Google Scholar] [CrossRef] [Green Version]
- Michael, C.A.; Labbate, M. Gene cassette transcription in a large integron-associated array. BMC Genet. 2010, 11, 82. [Google Scholar] [CrossRef] [Green Version]
- Mazel, D.; Dychinco, B.; Webb, V.A.; Davies, J. A Distinctive Class of Integron in the Vibrio cholerae Genome. Science 1998, 280, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Recchia, G.D.; Hall, R.M. Gene cassettes: A new class of mobile element. Microbiology 1995, 141, 3015–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biskri, L.; Bouvier, M.; Guérout, A.-M.; Boisnard, S.; Mazel, D. Comparative Study of Class 1 Integron and Vibrio cholerae Superintegron Integrase Activities. J. Bacteriol. 2005, 187, 1740–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemergut, D.R.; Robeson, M.S.; Kysela, R.F.; Martin, A.P.; Schmidt, S.K.; Knight, R. Insights and inferences about integron evolution from genomic data. BMC Genomics 2008, 9, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni Zhang, A.; Li, L.-G.; Ma, L.; Gillings, M.; Tiedje, J.M.; Zhang, T. Conserved phylogenetic distribution and limited antibiotic resistance of class 1 integrons revealed by assessing the bacterial genome and plasmid collection. Microbiome 2018, 6, 130. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yang, L.; Fu, J.; Yan, M.; Chen, D.; Zhang, L. Microbial pathogenicity and virulence mediated by integrons on Gram-positive microorganisms. Microb. Pathog. 2017, 111, 481–486. [Google Scholar] [CrossRef]
- Stalder, T.; Barraud, O.; Casellas, M.; Dagot, C.; Ploy, M.-C. Integron involvement in environmental spread of antibiotic resistance. Front Microbiol. 2012, 3, 119. [Google Scholar]
- Bikard, D.; Julié-Galau, S.; Cambray, G.; Mazel, D. The synthetic integron: An in vivo genetic shuffling device. Nucl. Acids Res. 2010, 38, e153. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [Green Version]
- Moura, A.; Soares, M.; Pereira, C.; Leitão, N.; Henriques, I.; Correia, A. INTEGRALL: A database and search engine for integrons, integrases and gene cassettes. Bioinformatics 2009, 25, 1096–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoval-Quintana, E.; Lauga, B.; Cagnon, C. Environmental integrons: The dark side of the integron world. Trends Microbiol. 2022, in press. [Google Scholar] [CrossRef]
- Pereira, M.B.; Wallroth, M.; Kristiansson, E.; Axelson-Fisk, M. HattCI: Fast and Accurate attC site Identification Using Hidden Markov Models. J. Comput. Biol. 2016, 23, 891–902. [Google Scholar]
- Ghaly, T.M.; Tetu, S.G.; Penesyan, A.; Qi, Q.; Rajabal, V.; Gillings, M.R. Discovery of integrons in Archaea: Platforms for cross-domain gene transfer. bioRxiv 2022. bioRxiv:2022.02.06.479319. [Google Scholar]
- Buongermino Pereira, M.; Österlund, T.; Eriksson, K.M. A comprehensive survey of integron-associated genes present in meta-genomes. BMC Genomics 2020, 21, 495. [Google Scholar]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar]
- Gibson, M.K.; Forsberg, K.; Dantas, G. Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J. 2015, 9, 207–216. [Google Scholar] [CrossRef]
- Feldgarden, M.; Brover, V.; Gonzalez-Escalona, N.; Frye, J.G.; Haendiges, J.; Haft, D.H.; Hoffmann, M.; Pettengill, J.B.; Prasad, A.B.; Tillman, G.E.; et al. AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci. Rep. 2021, 11, 12728. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L. The Pfam protein families database: Towards a more sus-tainable future. Nucl. Acids Res. 2016, 44, D279–D285. [Google Scholar]
- Perrin, A.; Rocha, E.P.C. PanACoTA: A modular tool for massive microbial comparative genomics. NAR Genomics Bioinform. 2021, 3, lqaa106. [Google Scholar]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Ghaly, T.M.; Tetu, S.G.; Gillings, M.R. Predicting the taxonomic and environmental sources of integron gene cassettes using structural and sequence homology of attC sites. Commun. Biol. 2021, 4, 946. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucl. Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haft, D.H.; DiCuccio, M.; Badretdin, A.; Brover, V.; Chetvernin, V.; O’Neill, K.; Li, W.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; et al. RefSeq: An update on prokaryotic genome annotation and curation. Nucl. Acids Res. 2018, 46, D851–D860. [Google Scholar] [CrossRef] [PubMed]
- Haudiquet, M.; Buffet, A.; Rendueles, O.; Rocha, E.P.C. Interplay between the cell envelope and mobile genetic elements shapes gene flow in populations of the nosocomial pathogen Klebsiella pneumoniae. PLoS Biol. 2021, 19, e3001276. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Lam, M.M.C.; Wick, R.R.; Watts, S.C.; Cerdeira, L.T.; Wyres, K.L.; Holt, K.E. A genomic surveillance framework and genotyping tool for Klebsiella pneumoniae and its related species complex. Nat. Commun. 2021, 12, 4188. [Google Scholar] [CrossRef]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2017, 35, 518–522. [Google Scholar] [CrossRef]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.-M. ARG-ANNOT, a New Bioinformatic Tool to Discover Antibiotic Resistance Genes in Bacterial Genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucl. Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Kleinheinz, K.A.; Joensen, K.G.; Larsen, M.V. Applying the ResFinder and VirulenceFinder web-services for easy identification of acquired antibiotic resistance and E. coli virulence genes in bacteriophage and prophage nucleotide sequences. Bacteriophage 2014, 4, e27943. [Google Scholar]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Chiarucci, A.; Bacaro, G.; Rocchini, D.; Fattorini, L. Discovering and rediscovering the sample-based rarefaction formula in the ecological literature. Commun. Ecol. 2008, 9, 121–123. [Google Scholar] [CrossRef]
- Waskom, M.L. Seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Smillie, C.; Pilar Garcillan-Barcia, M.; Victoria Francia, M.; Rocha, E.P.C.; de la Cruz, F. Mobility of Plasmids. Microbiol. Mol. Biol. Rev. 2010, 74, 434–452. [Google Scholar]
- Campos-Madueno, E.I.; Gmuer, C.; Risch, M.; Bodmer, T.; Endimiani, A. Characterisation of a new blaVIM-1-carrying IncN2 plasmid from an Enterobacter hormaechei subsp. steigerwaltii. J. Glob. Antimicrob. Resist. 2021, 24, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Daims, H.; Lebedeva, E.V.; Pjevac, P.; Han, P.; Herbold, C.; Albertsen, M.; Jehmlich, N.; Palatinszky, M.; Vierheilig, J.; Bulaev, A.; et al. Complete nitrification by Nitrospira bacteria. Nature 2015, 528, 504–509. [Google Scholar] [CrossRef] [PubMed]
- González-Torres, P.; Gabaldón, T. Genome Variation in the Model Halophilic Bacterium Salinibacter ruber. Front. Microbiol. 2018, 9, 1499. [Google Scholar] [CrossRef] [PubMed]
- Nešvera, J.; Hochmannová, J.; Pátek, M. An integron of class 1 is present on the plasmid pCG4 from Gram-positive bacterium Corynebacterium glutamicum. FEMS Microbiol. Lett. 1998, 169, 391–395. [Google Scholar] [CrossRef] [Green Version]
- Nandi, S.; Maurer, J.J.; Hofacre, C.; Summers, A.O. Gram-positive bacteria are a major reservoir of Class 1 antibiotic resistance integrons in poultry litter. Proc. Natl. Acad. Sci. USA 2004, 101, 7118–7122. [Google Scholar] [CrossRef] [Green Version]
- Paczosa, M.K.; Mecsas, J. Klebsiella pneumoniae: Going on the Offense with a Strong Defense. Microbiol. Mol. Biol. Rev. 2016, 80, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Poirel, L.; Le Thomas, I.; Naas, T.; Karim, A.; Nordmann, P. Biochemical Sequence Analyses of GES-1, a Novel Class A Extended-Spectrum β-Lactamase, and the Class 1 Integron In52 from Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2000, 44, 622–632. [Google Scholar] [CrossRef] [Green Version]
- Argimón, S.; AbuDahab, K.; Goater, R.J.E.; Fedosejev, A.; Bhai, J.; Glasner, C.; Feil, E.J.; Holden, M.T.G.; Yeats, C.A.; Grundmann, H.; et al. Microreact: Visualizing and sharing data for genomic epidemiology and phylogeography. Microb. Genomics 2016, 2, e000093. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, M.; Kumar, S.; Kapoor, R.K.; Virdi, J.S.; Gulati, P. Integrons in Enterobacteriaceae: Diversity, distribution and epidemiology. Int. J. Antimicrob. Agents 2018, 51, 167–176. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Néron, B.; Littner, E.; Haudiquet, M.; Perrin, A.; Cury, J.; Rocha, E.P.C. IntegronFinder 2.0: Identification and Analysis of Integrons across Bacteria, with a Focus on Antibiotic Resistance in Klebsiella. Microorganisms 2022, 10, 700. https://doi.org/10.3390/microorganisms10040700
Néron B, Littner E, Haudiquet M, Perrin A, Cury J, Rocha EPC. IntegronFinder 2.0: Identification and Analysis of Integrons across Bacteria, with a Focus on Antibiotic Resistance in Klebsiella. Microorganisms. 2022; 10(4):700. https://doi.org/10.3390/microorganisms10040700
Chicago/Turabian StyleNéron, Bertrand, Eloi Littner, Matthieu Haudiquet, Amandine Perrin, Jean Cury, and Eduardo P. C. Rocha. 2022. "IntegronFinder 2.0: Identification and Analysis of Integrons across Bacteria, with a Focus on Antibiotic Resistance in Klebsiella" Microorganisms 10, no. 4: 700. https://doi.org/10.3390/microorganisms10040700
APA StyleNéron, B., Littner, E., Haudiquet, M., Perrin, A., Cury, J., & Rocha, E. P. C. (2022). IntegronFinder 2.0: Identification and Analysis of Integrons across Bacteria, with a Focus on Antibiotic Resistance in Klebsiella. Microorganisms, 10(4), 700. https://doi.org/10.3390/microorganisms10040700