
Litter Management Strategies and Their Impact on the Environmental and Respiratory Microbiome Might Influence Health in Poultry

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
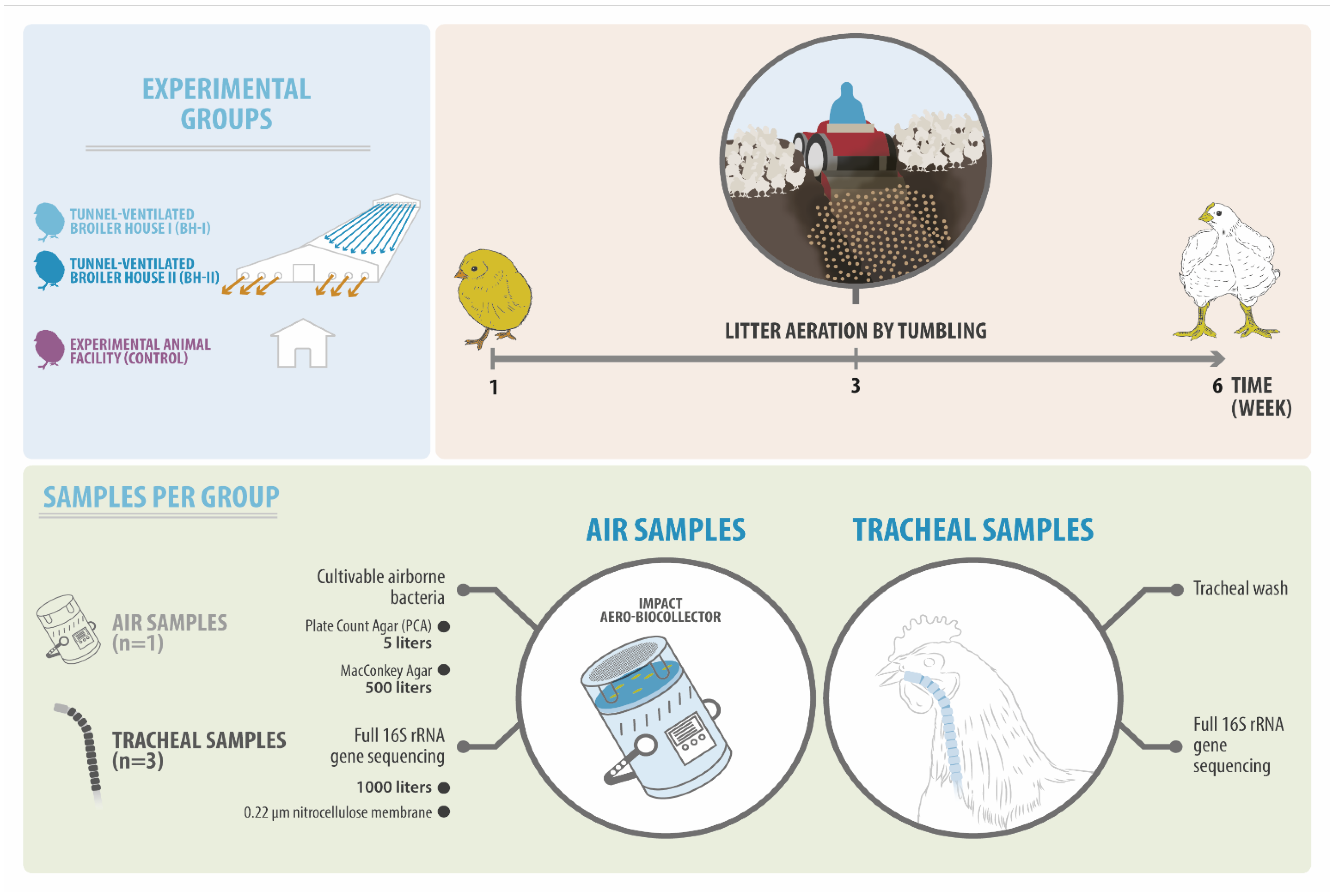
2.1. Experimental Design
2.2. Sample Collection
2.3. Sample Processing
2.4. Primers, 16S rRNA Gene Amplification, and Sequencing Process
2.5. Processing and OTU (Operational Taxonomic Units) Clustering
2.6. Phylogenetic Affiliation and OPU (Operational Phylogenetic Unit) Grouping
2.7. Diversity, Comparative, and Statistical Analysis
3. Results
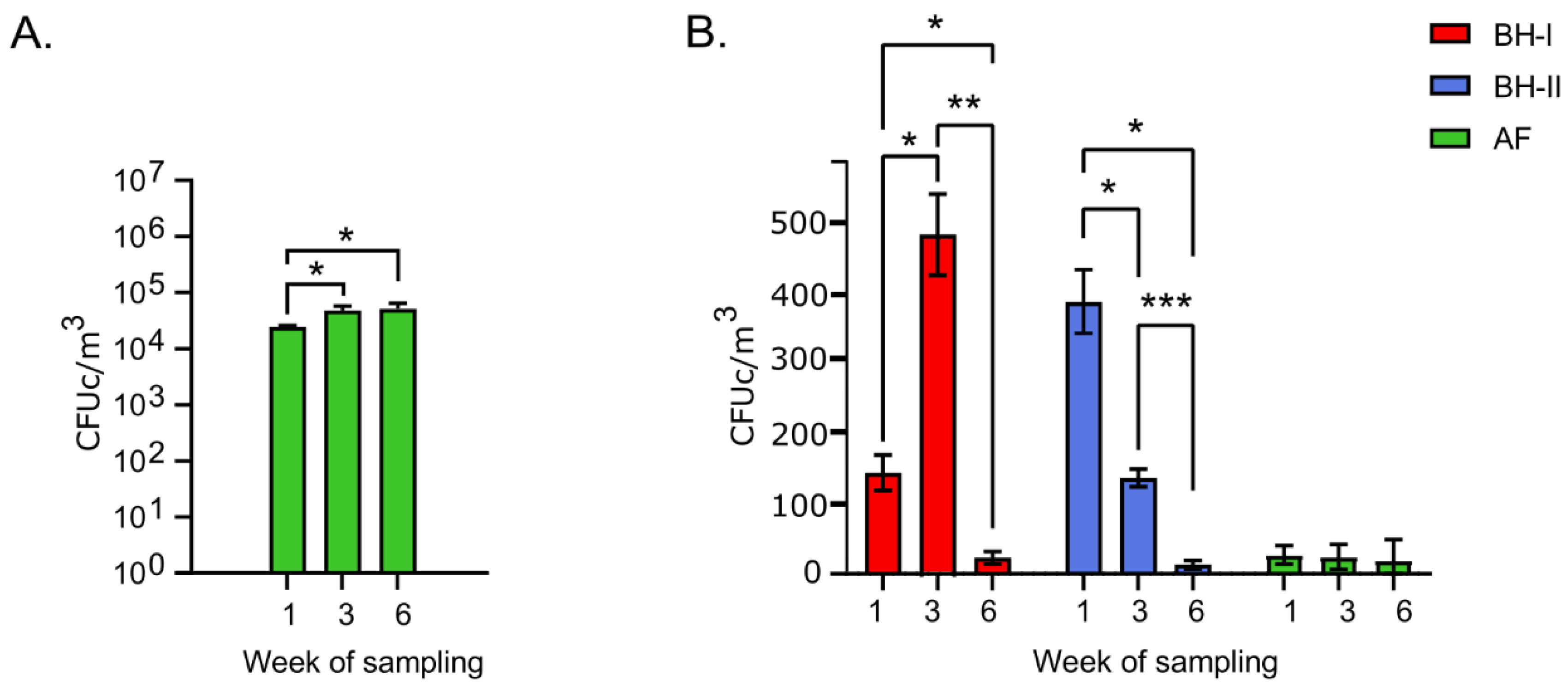
3.1. Viable Microorganisms in the Environment
3.2. 16S rRNA Gene Analysis of Microorganisms in the Environment and Host
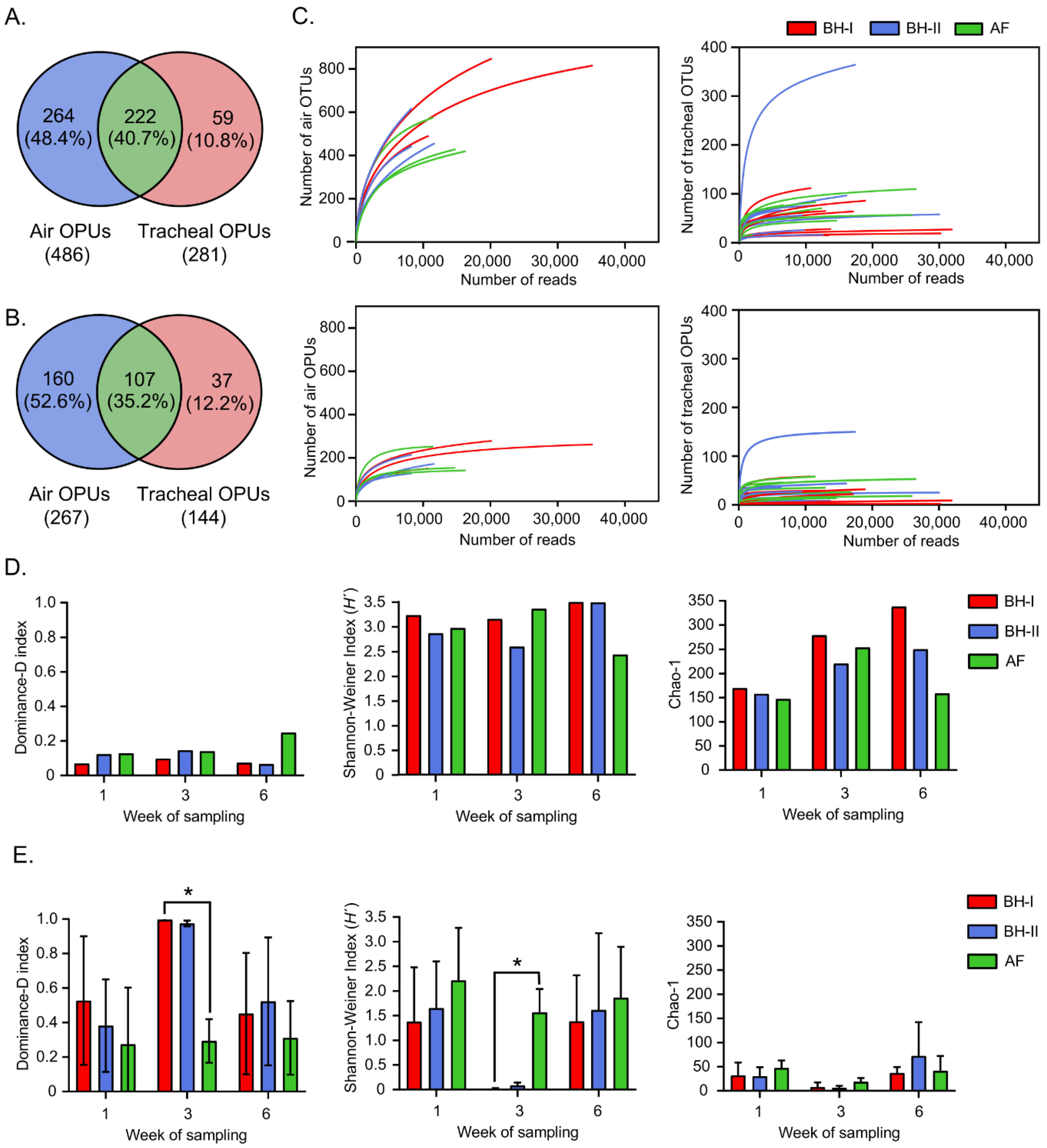
3.2.1. Samples, Sequences, and OPU Distribution
3.2.2. Tracheal Respiratory Samples Present Lower Diversity Than Environmental Air Samples
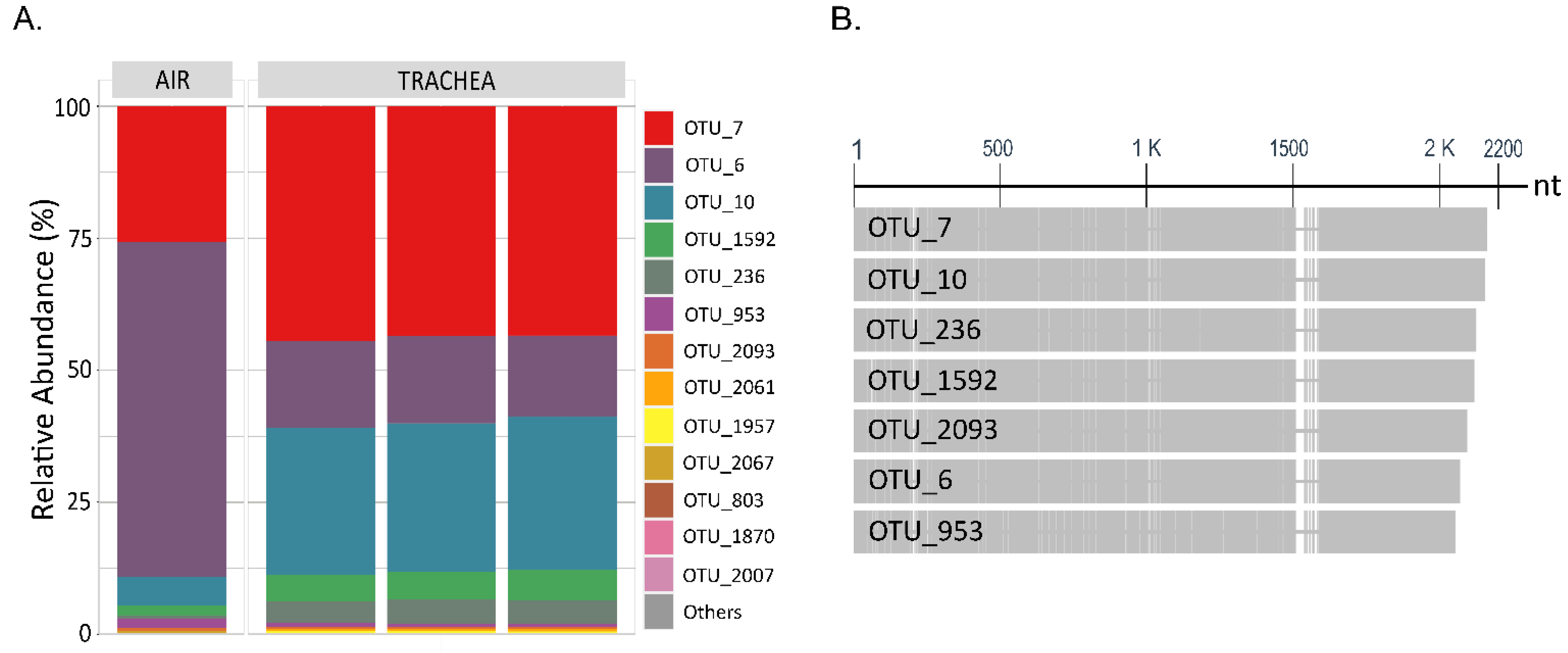
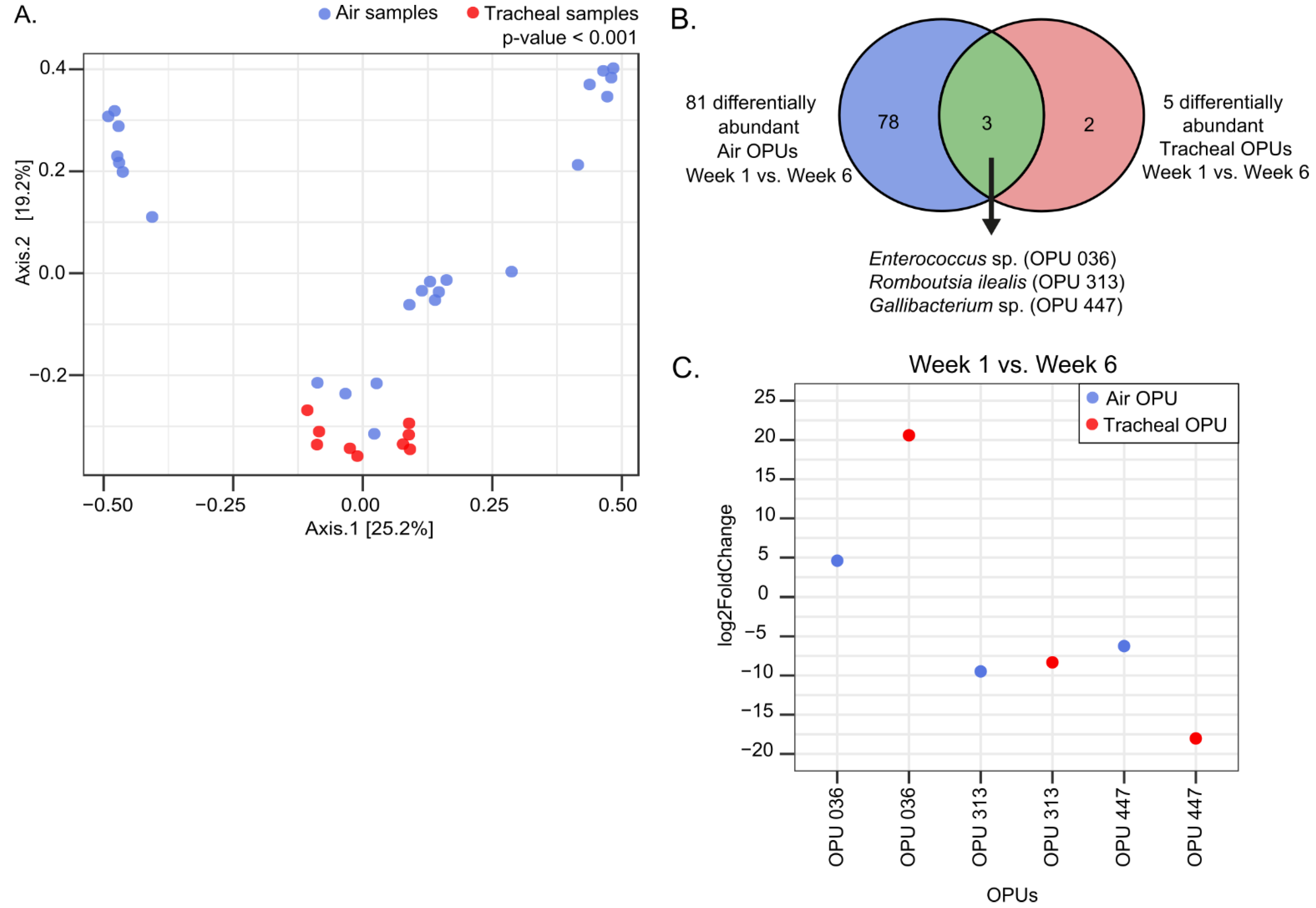
3.2.3. Environmental Air Shows a Distinctive Microbial Community
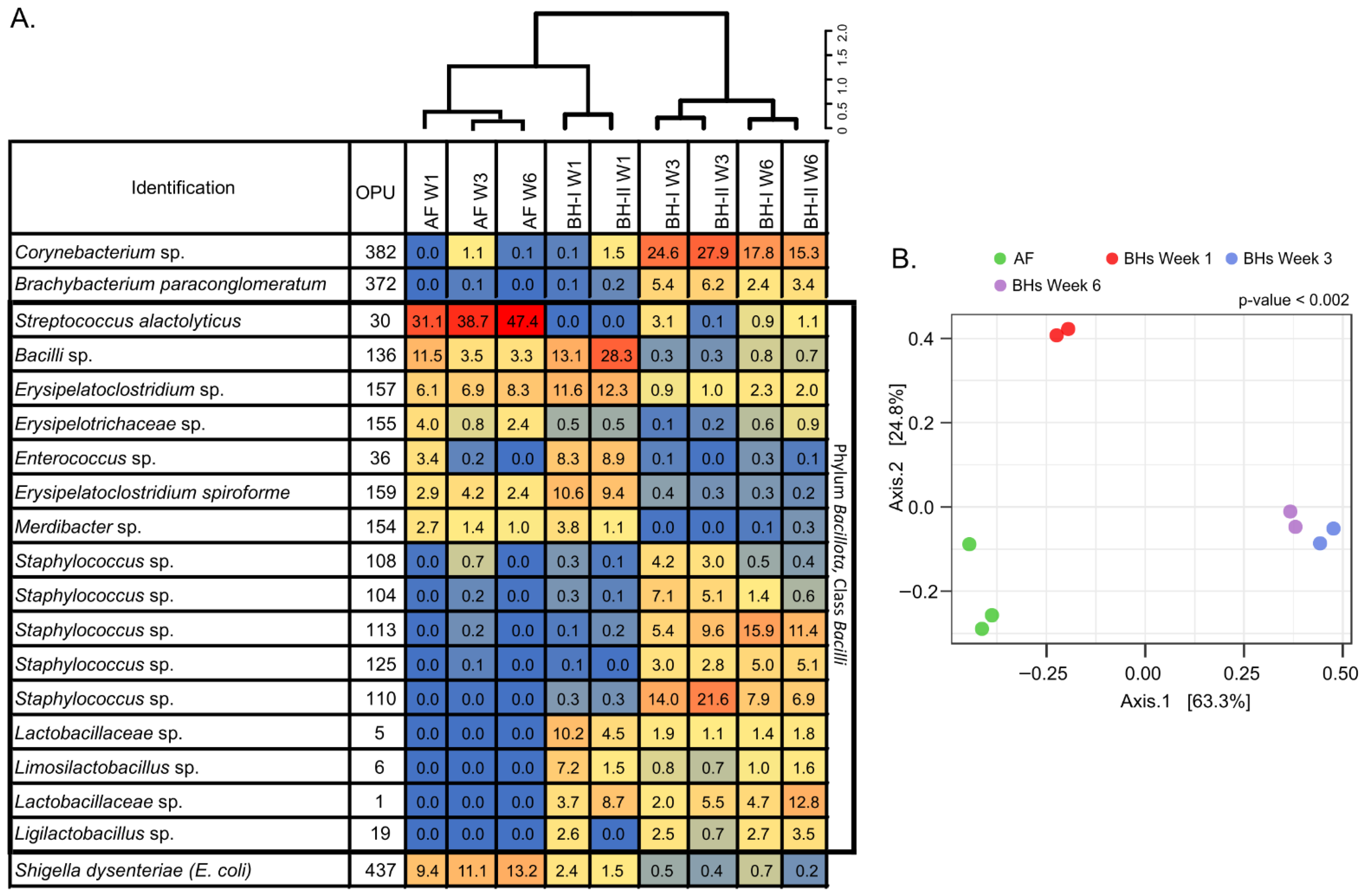
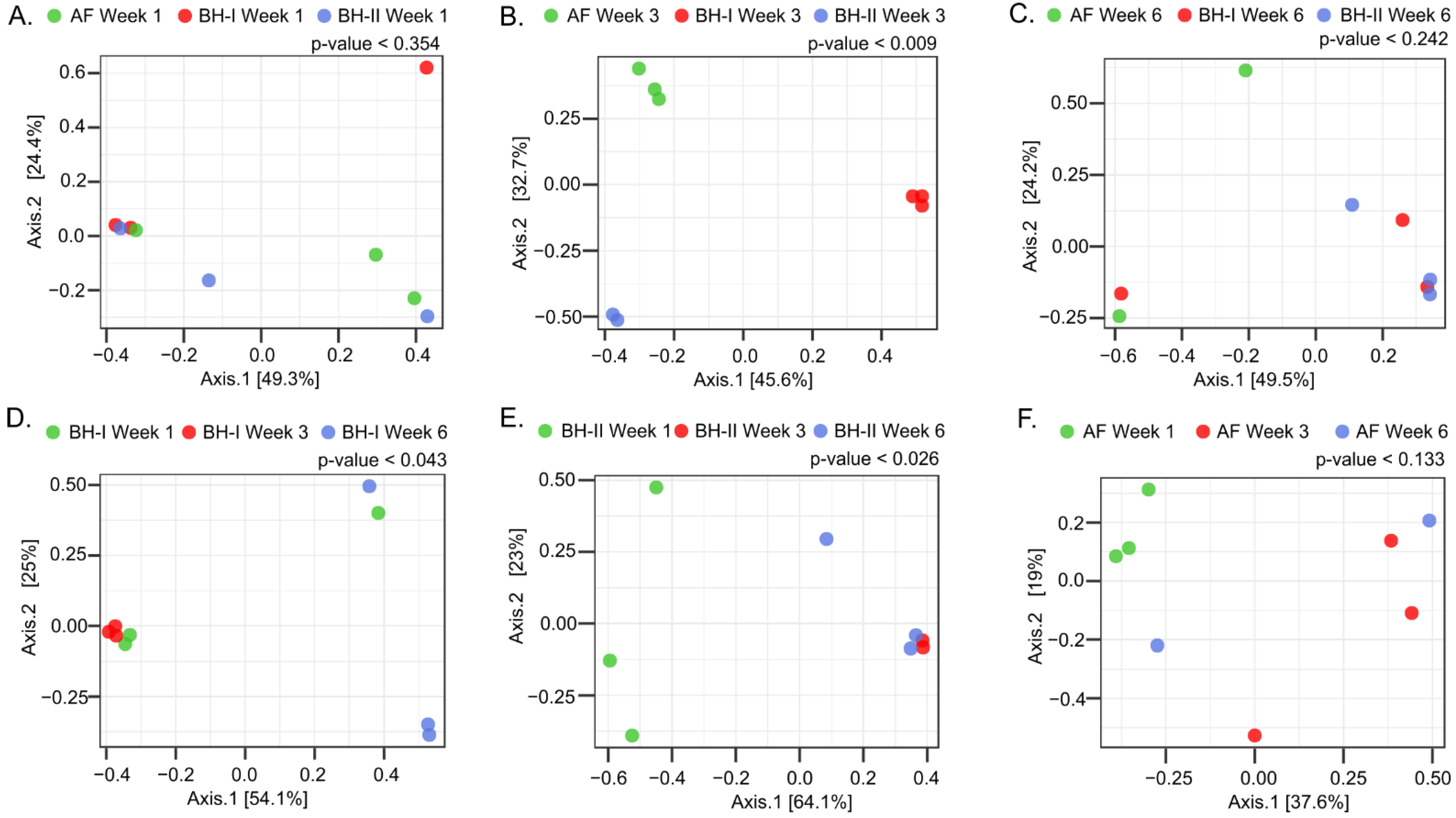
3.2.4. Tracheal Microbiota Composition Is Associated with Litter Management and Evolves in Broiler Houses
3.2.5. The Composition of Environmental Air and Tracheal Microbiota Are Different but Share Differentially Abundant Features over Time
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAO. Meat Market Review—Overview of Global Meat Market Developments in 2020, March 2021; FAO: Rome, Italy, 2021. [Google Scholar]
- Nolan, L.K.; Vaillancourt, J.P.; Barbieri, N.L.; Logue, C.M. Chapter 18: Colibacillosis. In Diseases of Poultry; Swayne, D.E., Boulianne, M., Logue, C.M., McDougald, L.R., Nair, V., Suarez, D.L., Eds.; Wiley-Blackwell: Ames, IA, USA, 2020; Volume 2, pp. 770–830. ISBN 978-1-119-37117-5. [Google Scholar]
- Jackwood, M.W. Review of Infectious Bronchitis Virus around the World. Avian Dis. 2012, 56, 634–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, J.G.; Patel, B.J.; Patel, S.S.; Raval, S.H.; Parmar, R.S.; Joshi, D.V.; Chauhan, H.C.; Chandel, B.S.; Patel, B.K. Metagenomic of Clinically Diseased and Healthy Broiler Affected with Respiratory Disease Complex. Data Brief 2018, 19, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Man, W.H.; de Steenhuijsen Piters, W.A.A.; Bogaert, D. The Microbiota of the Respiratory Tract: Gatekeeper to Respiratory Health. Nat. Rev. Microbiol. 2017, 15, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Mach, N.; Baranowski, E.; Nouvel, L.X.; Citti, C. The Airway Pathobiome in Complex Respiratory Diseases: A Perspective in Domestic Animals. Front. Cell. Infect. Microbiol. 2021, 11, 583600. [Google Scholar] [CrossRef]
- Shabbir, M.Z.; Malys, T.; Ivanov, Y.V.; Park, J.; Shabbir, M.A.B.; Rabbani, M.; Yaqub, T.; Harvill, E.T. Microbial Communities Present in the Lower Respiratory Tract of Clinically Healthy Birds in Pakistan. Poult. Sci. 2015, 94, 612–620. [Google Scholar] [CrossRef]
- Sohail, M.U.; Hume, M.E.; Byrd, J.A.; Nisbet, D.J.; Shabbir, M.Z.; Ijaz, A.; Rehman, H. Molecular Analysis of the Caecal and Tracheal Microbiome of Heat-Stressed Broilers Supplemented with Prebiotic and Probiotic. Avian Pathol. J. WVPA 2015, 44, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Johnson, T.J.; Youmans, B.P.; Noll, S.; Cardona, C.; Evans, N.P.; Karnezos, T.P.; Ngunjiri, J.M.; Abundo, M.C.; Lee, C.-W. A Consistent and Predictable Commercial Broiler Chicken Bacterial Microbiota in Antibiotic-Free Production Displays Strong Correlations with Performance. Appl. Environ. Microbiol. 2018, 84, e00362-18. [Google Scholar] [CrossRef] [Green Version]
- Mulholland, K.A.; Robinson, M.G.; Keeler, S.J.; Johnson, T.J.; Weber, B.W.; Keeler, C.L. Metagenomic Analysis of the Respiratory Microbiome of a Broiler Flock from Hatching to Processing. Microorganisms 2021, 9, 721. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, M.; Liu, Q.; Feng, J. The Alterations of Tracheal Microbiota and Inflammation Caused by Different Levels of Ammonia Exposure in Broiler Chickens. Poult. Sci. 2021, 100, 685–696. [Google Scholar] [CrossRef]
- Wheeler, E.; Casey, K.; Gates, R.; Xin, H.; Liang, Y.; Topper, P. Litter Management Strategies in Relation to Ammonia Emissions from Floor-Raised Birds. In Proceedings of the National Conference on Mitigating Air Emissions from Animal Feeding Operations. Exploring the Advantages Limitations, and Economics of Mitigation Technologies, Des Moines, IA, USA, 19–21 May 2008; Muhlbauer, E., Moody, L., Burns, R., Eds.; Iowa State University, College of Agriculture and Life Sciences: Des Moines, IA, USA, 2008; pp. 99–103, ISBN 978-0-9817781-0-5. [Google Scholar]
- Bodí, S.G.; Garcia, A.V.; García, S.V.; Orenga, C.M. Litter Aeration and Spread of Salmonella in Broilers. Poult. Sci. 2013, 92, 2005–2011. [Google Scholar] [CrossRef]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.-H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the Classification of Cultured and Uncultured Bacteria and Archaea Using 16S RRNA Gene Sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Pepper, C.-M.; Dunlop, M.W. Review of Litter Turning during a Grow-Out as a Litter Management Practice to Achieve Dry and Friable Litter in Poultry Production. Poult. Sci. 2021, 100, 101071. [Google Scholar] [CrossRef] [PubMed]
- Applebaum, J. What Is the Composition of the Avian Respiratory Microbiome? Thesis for the Degree of Bachelor of Science in Pre-Veterinary and Animal Biosciences. Bachelor’s Thesis, University of Delaware, Newark, DE, USA, 2014. [Google Scholar]
- Kashyap, S.K.; Maherchandani, S.; Kumar, N. Chapter 18—Ribotyping: A Tool for Molecular Taxonomy. In Animal Biotechnology; Verma, A.S., Singh, A., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 327–344. ISBN 978-0-12-416002-6. [Google Scholar]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, W.; Strunk, O.; Westram, R.; Richter, L.; Meier, H.; Yadhukumar; Buchner, A.; Lai, T.; Steppi, S.; Jobb, G.; et al. ARB: A Software Environment for Sequence Data. Nucleic Acids Res. 2004, 32, 1363–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruesse, E.; Peplies, J.; Glöckner, F.O. SINA: Accurate High-Throughput Multiple Sequence Alignment of Ribosomal RNA Genes. Bioinform. Oxf. Engl. 2012, 28, 1823–1829. [Google Scholar] [CrossRef] [PubMed]
- Vidal, R.; Ginard, D.; Khorrami, S.; Mora-Ruiz, M.; Munoz, R.; Hermoso, M.; Díaz, S.; Cifuentes, A.; Orfila, A.; Rosselló-Móra, R. Crohn Associated Microbial Communities Associated to Colonic Mucosal Biopsies in Patients of the Western Mediterranean. Syst. Appl. Microbiol. 2015, 38, 442–452. [Google Scholar] [CrossRef]
- Chamorro, N.; Montero, D.A.; Gallardo, P.; Farfán, M.; Contreras, M.; De la Fuente, M.; Dubois, K.; Hermoso, M.A.; Quera, R.; Pizarro-Guajardo, M.; et al. Landscapes and Bacterial Signatures of Mucosa-Associated Intestinal Microbiota in Chilean and Spanish Patients with Inflammatory Bowel Disease. Microb. Cell Graz Austria 2021, 8, 223–238. [Google Scholar] [CrossRef]
- Hammer, O.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4, 4–9. [Google Scholar]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for Comprehensive Statistical, Functional, and Meta-Analysis of Microbiome Data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A Web-Based Tool for the Analysis of Sets through Venn Diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, P.; Izquierdo, M.; Vidal, R.M.; Chamorro-Veloso, N.; Rosselló-Móra, R.; O’Ryan, M.; Farfán, M.J. Distinctive Gut Microbiota Is Associated with Diarrheagenic Escherichia Coli Infections in Chilean Children. Front. Cell. Infect. Microbiol. 2017, 7, 424. [Google Scholar] [CrossRef] [Green Version]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef] [Green Version]
- Dunlop, M.W.; Moss, A.F.; Groves, P.J.; Wilkinson, S.J.; Stuetz, R.M.; Selle, P.H. The Multidimensional Causal Factors of “wet Litter” in Chicken-Meat Production. Sci. Total Environ. 2016, 562, 766–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aspee, F.; (Agrícola Ariztía Ltda., Santiago, Chile). Personal Communication. 2022.
- García-Solache, M.; Rice, L.B. The Enterococcus: A Model of Adaptability to Its Environment. Clin. Microbiol. Rev. 2019, 32, e00058-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logue, C.M.; Abdul-Aziz, T. Chapter 23: Other Bacterial Diseases. In Diseases of Poultry; Swayne, D.E., Boulianne, M., Logue, C.M., McDougald, L.R., Nair, V., Suarez, D.L., Eds.; Wiley-Blackwell: Ames, IA, USA, 2020; Volume 2, pp. 1047–1085. ISBN 978-1-119-37117-5. [Google Scholar]
- Narasinakuppe Krishnegowda, D.; Dhama, K.; Kumar Mariappan, A.; Munuswamy, P.; Iqbal Yatoo, M.; Tiwari, R.; Karthik, K.; Bhatt, P.; Reddy, M.R. Etiology, Epidemiology, Pathology, and Advances in Diagnosis, Vaccine Development, and Treatment of Gallibacterium Anatis Infection in Poultry: A Review. Vet. Q. 2020, 40, 16–34. [Google Scholar] [CrossRef] [Green Version]
- Maki, J.J.; Nielsen, D.W.; Looft, T. Complete Genome Sequence and Annotation for Romboutsia Sp. Strain CE17. Microbiol. Resour. Announc. 2020, 9, e00382-20. [Google Scholar] [CrossRef]
- Schouler, C.; Schaeffer, B.; Brée, A.; Mora, A.; Dahbi, G.; Biet, F.; Oswald, E.; Mainil, J.; Blanco, J.; Moulin-Schouleur, M. Diagnostic Strategy for Identifying Avian Pathogenic Escherichia Coli Based on Four Patterns of Virulence Genes. J. Clin. Microbiol. 2012, 50, 1673–1678. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivulic, D.; Rossello-Mora, R.; Viver, T.; Montero, D.A.; Vidal, S.; Aspee, F.; Hidalgo, H.; Vidal, R. Litter Management Strategies and Their Impact on the Environmental and Respiratory Microbiome Might Influence Health in Poultry. Microorganisms 2022, 10, 878. https://doi.org/10.3390/microorganisms10050878
Ivulic D, Rossello-Mora R, Viver T, Montero DA, Vidal S, Aspee F, Hidalgo H, Vidal R. Litter Management Strategies and Their Impact on the Environmental and Respiratory Microbiome Might Influence Health in Poultry. Microorganisms. 2022; 10(5):878. https://doi.org/10.3390/microorganisms10050878
Chicago/Turabian StyleIvulic, Dinka, Ramon Rossello-Mora, Tomeu Viver, David A. Montero, Sonia Vidal, Francisco Aspee, Héctor Hidalgo, and Roberto Vidal. 2022. "Litter Management Strategies and Their Impact on the Environmental and Respiratory Microbiome Might Influence Health in Poultry" Microorganisms 10, no. 5: 878. https://doi.org/10.3390/microorganisms10050878
APA StyleIvulic, D., Rossello-Mora, R., Viver, T., Montero, D. A., Vidal, S., Aspee, F., Hidalgo, H., & Vidal, R. (2022). Litter Management Strategies and Their Impact on the Environmental and Respiratory Microbiome Might Influence Health in Poultry. Microorganisms, 10(5), 878. https://doi.org/10.3390/microorganisms10050878