
Genomic and Physiological Characterization of Metabacillus flavus sp. nov., a Novel Carotenoid-Producing Bacilli Isolated from Korean Marine Mud

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strain Isolation and Cultivation
2.2. DNA Extraction and Phylogenetic Analysis
2.3. Phenotypic Characterization
2.4. Chemotaxonomic Characterization
2.5. Whole-Genome Sequencing and Verification of Authenticity of the Genome Assembly
2.6. Genome Annotation and Phylogenomic and Comparative Genomic Analysis
2.7. Pan-Genomic Analysis
2.8. Pigment Extraction and Analysis
3. Results and Discussion
3.1. Morphological, Physiological, and Biochemical Characterization and Discrimination
3.2. Chemotaxonomic Characterization
3.3. Whole-Genome Sequencing and Verification of Authenticity of the Genome Assembly
3.4. 16S rRNA Gene Phylogeny
3.5. Genome-Derived Features and Comparative Genomic Analysis
3.6. Pan-Genomic Analysis of the Genus Metabacillus including Strain KIGAM252T
3.7. Identification of Carotenoid in the Strain KIGAM252T
4. Conclusions
Description of Metabacillus flavus sp. nov.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patel, S.; Gupta, R.S. A phylogenomic and comparative genomic framework for resolving the polyphyly of the genus Bacillus: Proposal for six new genera of Bacillus species, Peribacillus gen. nov., Cytobacillus gen. nov., Mesobacillus gen. nov., Neobacillus gen. nov., Metabacillus gen. nov. and Alkalihalobacillus gen. nov. Int. J. Syst. Evol. Microbiol. 2020, 70, 406–438. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.; Ahmed, I.; Kudo, T.; Iqbal, M.; Lee, Y.J.; Fujiwara, T.; Ohkuma, M. A heavy metal tolerant novel bacterium, Bacillus malikii sp. nov., isolated from tannery effluent wastewater. Antonie Van Leeuwenhoek 2015, 108, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Balcázar, J.L.; Pintado, J.; Planas, M. Bacillus galliciensis sp. nov., isolated from faeces of wild seahorses (Hippocampus guttulatus). Int. J. Syst. Evol. Microbiol. 2010, 60, 892–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.H.; Tian, X.R.; Ruan, Y.; Yang, L.L.; He, Z.Q. Bacillus crassostreae sp. nov., isolated from an oyster (Crassostrea hongkongensis). Int. J. Syst. Evol. Microbiol. 2015, 65, 1561–1566. [Google Scholar] [CrossRef] [Green Version]
- Dooren, D.; de Jong, L.E. Über Bacillus fastidiosus. Zentrbl. Bakt. Abt. 1929, 2, 334–358. [Google Scholar]
- Gupta, V.; Singh, P.K.; Korpole, S.; Tanuku, N.R.S.; Pinnaka, A.K. Bacillus mangrovi sp. nov., isolated from a sediment sample from a mangrove forest. Int. J. Syst. Evol. Microbiol. 2017, 67, 2219–2224. [Google Scholar] [CrossRef]
- Ko, K.S.; Oh, W.S.; Lee, M.Y.; Lee, J.H.; Lee, H.; Peck, K.R.; Lee, N.Y.; Song, J.H. Bacillus infantis sp. nov. and Bacillus idriensis sp. nov., isolated from a patient with neonatal sepsis. Int. J. Syst. Evol. Microbiol. 2006, 56, 2541–2544. [Google Scholar] [CrossRef]
- Kwon, S.W.; Lee, S.Y.; Kim, B.Y.; Weon, H.Y.; Kim, J.B.; Go, S.J.; Lee, G.B. Bacillus niabensis sp. nov., isolated from cotton-waste composts for mushroom cultivation. Int. J. Syst. Evol. Microbiol. 2007, 57, 1909–1913. [Google Scholar] [CrossRef]
- Mehrshad, M.; Amoozegar, M.A.; Didari, M.; Bagheri, M.; Fazeli, S.A.S.; Schumann, P.; Spröer, C.; Sánchez-Porro, C.; Ventosa, A. Bacillus halosaccharovorans sp. nov., a moderately halophilic bacterium from a hypersaline lake. Int. J. Syst. Evol. Microbiol. 2013, 63, 2776–2781. [Google Scholar] [CrossRef]
- Parag, B.; Sasikala, C.; Ramana, C.V. Bacillus endolithicus sp. nov., isolated from pebbles. Int. J. Syst. Evol. Microbiol. 2015, 65, 4568–4573. [Google Scholar] [CrossRef]
- Singh, H.; Kaur, M.; Kaur, L.; Sharma, S.; Mishra, S.; Tunuku, N.R.S.; Pinnaka, A.K. Bacillus lacus sp. nov., isolated from a water sample of a salt lake in India. Int. J. Syst. Evol. Microbiol. 2018, 68, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Suresh, K.; Prabagaran, S.R.; Sengupta, S.; Shivaji, S. Bacillus indicus sp. nov., an arsenic-resistant bacterium isolated from an aquifer in West Bengal, India. Int. J. Syst. Evol. Microbiol. 2004, 54, 1369–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.I.; Zhang, J.; Sun, L. Bacillus iocasae sp. nov., isolated from Pacmanus hydrothermal field, Manus Basin. Int. J. Syst. Evol. Microbiol. 2017, 67, 3547–3552. [Google Scholar] [CrossRef] [PubMed]
- Wieser, M.; Worliczek, H.; Kämpfer, P.; Busse, H.J. Bacillus herbersteinensis sp. nov. Int. J. Syst. Evol. Microbiol. 2005, 55, 2119–2123. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.H.; Oh, T.K. Bacillus litoralis sp. nov., isolated from a tidal flat of the Yellow Sea in Korea. Int. J. Syst. Evol. Microbiol. 2005, 55, 1945–1948. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Son, J.S.; Hwang, Y.J.; Shin, J.H.; Ghim, S.Y. Metabacillus elymi sp. nov., isolated from the Rhizosphere of Elymus tsukushiensis, a plant native to the Dokdo Islands, Republic of Korea. Antonie Van Leeuwenhoek 2021, 114, 1709–1719. [Google Scholar] [CrossRef]
- Mao, H.; Wei, Y.; Gao, Y.; Pei, J.; Zhang, Y.; Fang, J. Metabacillus sediminilitoris sp. nov., a marine bacterium isolated from a tidal sediment. Int. J. Syst. Evol. Microbiol. 2020, 70, 5211–5216. [Google Scholar] [CrossRef]
- Britton, G.; Liaaen-Jensen, S.; Pfander, H. Carotenoids: Handbook; Birkhäuser: Basel, Switzerland, 2012. [Google Scholar]
- Rodriguez-Villalon, A.; Perez-Gil, J.; Rodriguez-Concepcion, M. Carotenoid accumulation in bacteria with enhanced supply of isoprenoid precursors by upregulation of exogenous or endogenous pathways. J. Biotechnol. 2008, 135, 78–84. [Google Scholar] [CrossRef]
- Heider, S.A.; Peters-Wendisch, P.; Wendisch, V.F.; Beekwilder, J.; Brautaset, T. Metabolic engineering for the microbial production of carotenoids and related products with a focus on the rare C50 carotenoids. Appl. Microbiol. Biotechnol. 2014, 98, 4355–4367. [Google Scholar] [CrossRef]
- Wang, F.; Jiang, J.G.; Chen, Q. Progress on molecular breeding and metabolic engineering of biosynthesis pathways of C30, C35, C40, C45, C50 carotenoids. Biotechnol. Adv. 2007, 25, 211–222. [Google Scholar] [CrossRef]
- Duc, L.H.; Fraser, P.; Cutting, S.M. Carotenoids present in halotolerant Bacillus spore formers. FEMS Microbiol. Lett. 2006, 255, 215–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korumilli, T.; Mishra, S. Carotenoid production by Bacillus clausii using rice powder as the sole substrate: Pigment analyses and optimization of key production parameters. J. Biochem. Technol. 2014, 5, 788–794. [Google Scholar]
- Perez-Fons, L.; Steiger, S.; Khaneja, R.; Bramley, P.M.; Cutting, S.M.; Sandmann, G.; Fraser, P.D. Identification and the developmental formation of carotenoid pigments in the yellow/orange Bacillus spore-formers. Biochem. Biophys. Acta. 2011, 1811, 177–185. [Google Scholar] [CrossRef]
- Osawa, A.; Iki, K.; Sandmann, G.; Shindo, K. Isolation and identification of 4, 4′-diapolycopene-4,4′-dioic acid produced by Bacillus firmus GB1 and its singlet oxygen quenching activity. J. Oleo. Sci. 2013, 62, 955–960. [Google Scholar] [CrossRef]
- Steiger, S.; Perez-Fons, L.; Fraser, P.D.; Sandmann, G. Biosynthesis of a novel C30 carotenoid in Bacillus firmus isolates. J. Appl. Microbiol. 2012, 113, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Jannasch, H.W.; Taylor, C.D. Deep-sea microbiology. Annu. Rev. Microbiol. 1984, 38, 487–514. [Google Scholar] [CrossRef] [PubMed]
- Kato, C.; Sato, T.; Horikoshi, K. Isolation and properties of barophilic and barotolerant bacteria from deep-sea mud samples. Biodivers. Conserv. 1995, 4, 1–9. [Google Scholar] [CrossRef]
- Takami, H.; Kobata, K.; Nagahama, T.; Kobayashi, H.; Inoue, A.; Horikoshi, K. Biodiversity in deep-sea sites located near the south part of Japan. Extremophiles 1999, 3, 97–102. [Google Scholar] [CrossRef]
- Cheng, T.H.; Ismail, N.; Kamaruding, N.; Saidin, J.; Danish-Daniel, M. Industrial enzymes-producing marine bacteria from marine resources. Biotechnol. Rep. 2020, 27, e00482. [Google Scholar] [CrossRef]
- Jahromi, S.T.; Barzkar, N. Future direction in marine bacterial agarases for industrial applications. Appl. Microbiol. Biotechnol. 2018, 102, 6847–6863. [Google Scholar] [CrossRef]
- Porras, M.A.; Witale, C.; Villar, M.A.; Cubitto, M.A. Bioconversion of glycerol to poly (HB-co-HV) copolymer in an inexpensive medium by a Bacillus megaterium strain isolated from marine sediments. J. Environ. Chem. Eng. 2017, 5, 1–9. [Google Scholar] [CrossRef]
- Lane, D.J. 16S/23S rRNA sequencing. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E.G., Ed.; John Wiley and Sons: New York, NY, USA, 1991; pp. 115–148. [Google Scholar]
- Roh, S.W.; Sung, Y.; Nam, Y.D.; Chang, H.W.; Kim, K.H.; Yoon, J.H.; Jeon, C.O.; Oh, H.M.; Bae, J.W. Arthrobacter soli sp. nov., a novel bacterium isolated from wastewater reservoir sediment. J. Microbiol. 2008, 46, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Thombre, R.S.; Shinde, V.D.; Oke, R.S.; Dhar, S.K.; Shouche, Y.S. Biology and survival of extremely halophilic archaeon Haloarcula marismortui RR12 isolated from Mumbai salterns, India in response to salinity stress. Sci. Rep. 2016, 6, 25642. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbour-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Kluge, A.G.; Farris, J.S. Quantitative phyletics and the evolution of anurans. Syst. Biol. 1969, 18, 1–32. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Tittsler, R.P.; Sandholzer, L.A. The use of semi-solid agar for the detection of bacterial motility. J. Bacteriol. 1936, 31, 575–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, H.J. Microbiological Applications: A Laboratory Manual in General Microbiology; McGraw-Hill Company Inc.: Massachusetts, MA, USA, 2002; p. 432. [Google Scholar]
- Bauer, A.W.; Kirby, M.M.; Sherris, J.C.; Truck, M. Antibiotic susceptibility testing by a standardized single disk method. Am. J. Clin. Pathol. 1966, 45, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.T. Single derivatization method for routine analysis of bacterial whole-cell fatty acid methyl esters, including hydroxy acids. J. Clin. Microbiol. 1982, 16, 584–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasser, M. Identification of bacteria by gas chromatography of cellular fatty acids. USFCC Newsl. 1990, 20, 1–6. [Google Scholar]
- Collins, M.D.; Jones, D. Distribution of isoprenoid quinone structural types in bacteria and their taxonomic implication. Microbiol. Rev. 1981, 45, 316–354. [Google Scholar] [CrossRef]
- Minnikin, D.E.; O’Donnell, A.G.; Goodfellow, M. An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J. Microbiol. Methods 1984, 2, 233–241. [Google Scholar] [CrossRef]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Chalita, M.; Ha, S.M.; Na, S.I.; Yoon, S.H.; Chun, J. ContEst16S: An algorithm that identifies contaminated prokaryotic genomes using 16S RNA gene sequences. Int. J. Syst. Evol. Microbiol. 2017, 67, 2053–2057. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucl. Acids. Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic. Acids. Res. 2009, 47, W81–W87. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Kim, Y.O.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Kim, D.; Park, S.; Chun, J. Introducing EzAAI: A pipeline for high throughput calculations of prokaryotic average amino acid identity. J. Microbiol. 2021, 59, 476–480. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [Green Version]
- Auch, A.F.; von Jan, M.; Klenk, H.P.; Göker, M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic. Sci. 2010, 2, 117–134. [Google Scholar] [CrossRef] [Green Version]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [Green Version]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA—An ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Khaneja, R.; Perez-Fons, L.; Fakhry, S.; Baccigalupi, L.; Steiger, S.; To, E.; Sandmann, G.; Dong, T.C.; Ricca, E.; Fraser, P.D.; et al. Carotenoids found in Bacillus. J. Appl. Microbiol. 2010, 108, 1889–1902. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.W.; de Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef]
- Köcher, S.; Breitenbach, J.; Müller, V.; Sandmann, G. Structure, function and biosynthesis of carotenoids in the moderately halophilic bacterium Halobacillus halophilus. Arch. Microbiol. 2009, 191, 95–104. [Google Scholar] [CrossRef]
- Zhu, S.; Hegemann, J.D.; Fage, C.D.; Zimmermann, M.; Xie, X.; Linne, U.; Marahiel, M.A. Insights into the unique phosphorylation of the lasso peptide paeninodin. J. Biol. Chem. 2016, 291, 13662–13678. [Google Scholar] [CrossRef] [Green Version]
- Steiger, S.; Perez-Fons, L.; Cutting, S.M.; Fraser, P.D.; Sandmann, G. Annotation and functional assignment of the genes for the C30 carotenoid pathways from the genomes of two bacteria: Bacillus indicus and Bacillus firmus. Microbiology 2015, 161, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Raisig, A.; Sandmann, G. Functional properties of diapophytoene and related desaturases of C-30 and C-40 carotenoid biosynthetic pathways. Biochim. Biophys. Acta 2001, 1533, 164–170. [Google Scholar] [CrossRef]
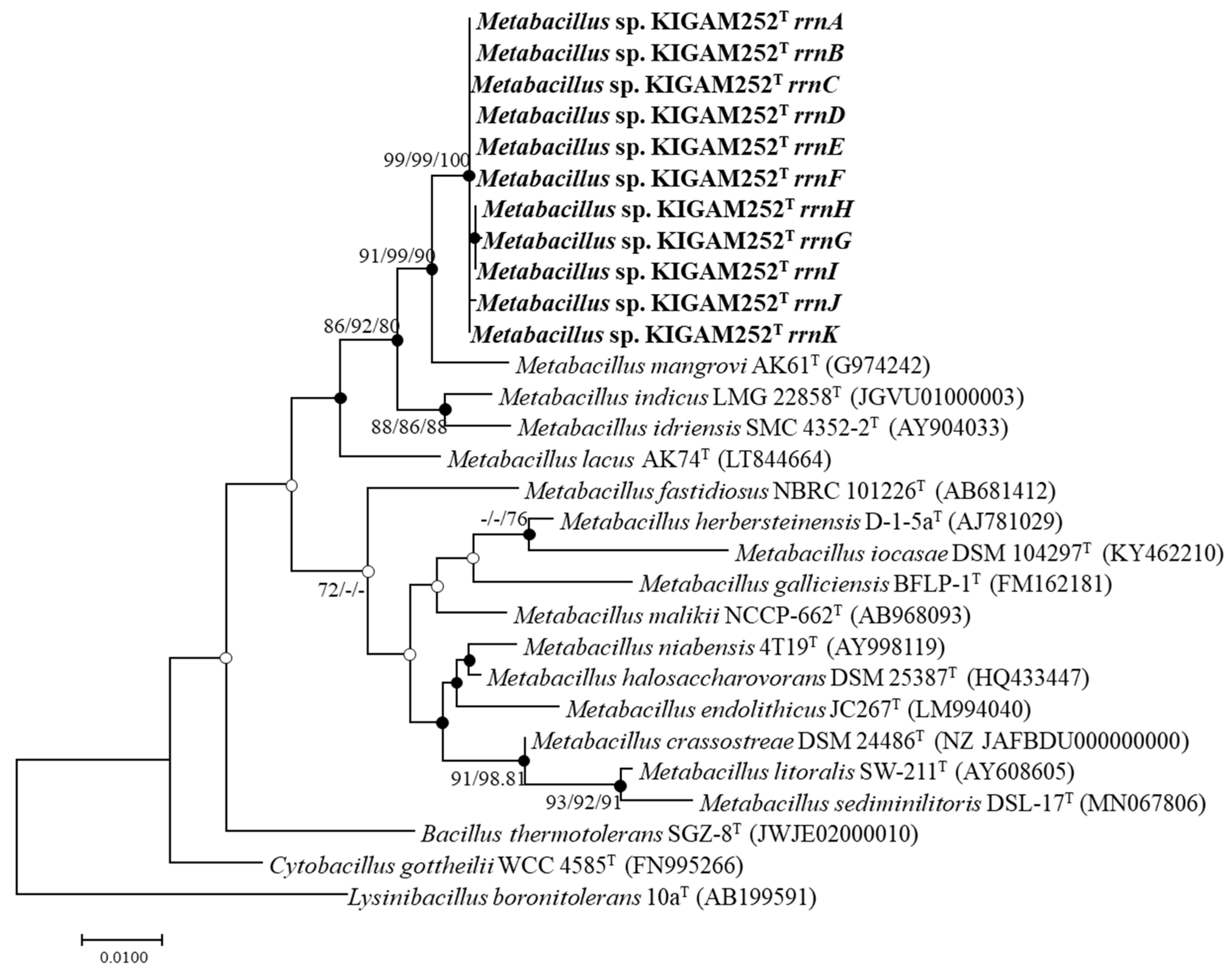

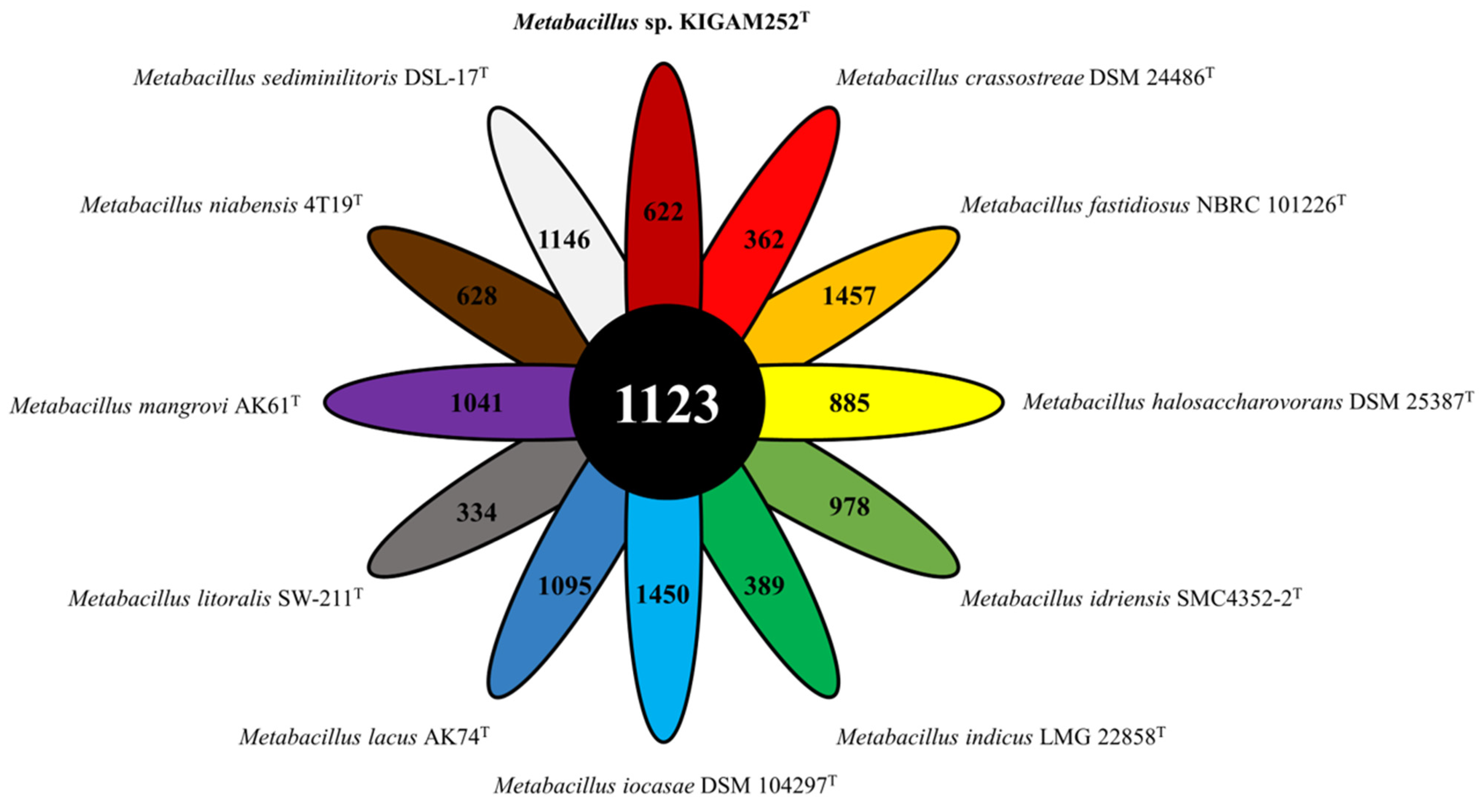


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Temperature range (°C) (optimum) | 10–35 (30) * | 15–42 a | 15–37 b | 15–45 c |
| NaCl range (%, w/v) (optimum) | 0–6 (3) * | 0–3 a | 0–2 b | 0–3 c |
| pH range for growth (optimum) | 6.0–10.0 (7.0) * | 6.0–9.0 a | 6.0–7.0 b | 5.5–9.5 c |
| Catalase * | + | − | + | − |
| Oxidase * | + | − | + | + |
| Hydrolysis of * | ||||
| Casein | + | − | + | + |
| Gelatin | + | − | − | − |
| Starch | − | + | + | + |
| Methyl red * | − | − | + | − |
| H2S production * | + | − | − | − |
| Esterase (C4) | − | + | + | + |
| Lipase (C14) | − | + | − | − |
| Valine arylamidase | − | + | + | + |
| Cystine arylamidase | − | + | − | − |
| Trypsin | + | − | − | − |
| α-Chymotrypsin | + | − | + | + |
| Acid phosphatase | − | − | − | + |
| β-glucosidase | − | + | − | − |
| D-Ribose | + | − | − | + |
| D-Xylose | − | − | − | + |
| D-Mannose | − | − | + | + |
| Inositol | − | − | − | + |
| D-Mannitol | + | + | − | + |
| D-Sorbitol | − | − | − | + |
| Methyl-α-D-mannoside | + | − | + | − |
| Methyl-α-D-glucoside | + | − | + | + |
| N-acetylglucosamine | + | − | + | + |
| Amygdalin | + | − | + | + |
| Arbutin | + | − | − | + |
| Salicin | + | − | + | + |
| D-Cellobiose | + | − | + | − |
| D-Lactose | − | − | + | + |
| D-Melibiose | − | + | + | + |
| Sucrose | + | + | − | + |
| D-Trehalose | + | + | − | + |
| Inulin | − | + | + | − |
| D-Melezitose | + | − | − | + |
| Starch | − | + | + | + |
| Glycogen | − | + | + | + |
| Gentiobiose | − | − | + | − |
| D-Turanose | − | − | + | + |
| 2-Ketogluconate | − | − | + | + |
| Saturated | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| C10:0 | ||||
| C12:0 | 0.47 | 0.10 | 0.04 | 0.16 |
| C14:0 | 0.21 | 0.12 | ND | 0.15 |
| C16:0 | 1.50 | 3.00 | 0.89 | 0.62 |
| C17:0 | 8.14 | 9.49 | 11.02 | 3.55 |
| C18:0 | ND | ND | 0.09 | ND |
| Unsaturated | ||||
| C16:1 ω7c alcohol | ND | 2.04 | 2.36 | 3.78 |
| C16:1 ω11c | ND | 5.75 | 8.71 | 0.73 |
| C18:1 ω9c | ND | ND | 0.22 | ND |
| Branched-chain fatty acid | ||||
| C11:0 iso | ND | 0.22 | ND | 0.09 |
| C13:0 iso | ND | 0.12 | ND | ND |
| C14:0 iso | 7.81 | 3.81 | 4.04 | 6.88 |
| C15:0 iso | 25.61 | 39.78 | 19.29 | 21.92 |
| C16:0 iso | 9.78 | 6.34 | 7.59 | 10.35 |
| C17:0 iso | 4.67 | 4.76 | 6.44 | 5.54 |
| C17:1 iso ω10c | ND | 2.16 | 2.41 | 0.28 |
| C18:0 iso | ND | ND | 0.20 | 0.32 |
| C19:0 iso | ND | ND | 0.08 | ND |
| C11:0 anteiso | ND | 0.18 | ND | 0.19 |
| C15:0 anteiso | 35.68 | 15.45 | 23.75 | 35.35 |
| C17:0 anteiso | 5.79 | 5.44 | 10.15 | 9.09 |
| Summed feature | ||||
| 3; C16:1 ω7c/C16:1 ω6c | ND | ND | 0.17 | ND |
| 4; C17:1 iso /C17:1 anteiso B | ND | 0.99 | 1.48 | 0.74 |
| Attribute | Characteristics |
|---|---|
| Sequencing platforms | PacBio |
| Assembler | FLYE v. 2.7 |
| Genome coverage | 292.0× |
| Assembly status | Complete |
| Assembly size (bp) | 4,302,488 bp |
| G + C content (%) | 43.8 |
| N50 | 4,026,853 |
| Total contigs | 2 |
| Chromosome | 2 |
| G + C content (%) | 43.78 (JAGVRK010000001) 52.19 (JAGVRK010000002) |
| Total genes | 4092 |
| Total CDS | 3964 |
| Coding genes | 3898 |
| Pseudo genes | 66 |
| RNAs | 128 |
| -rRNA genes (5S, 16S, 23S) | 11, 11, 12 |
| -tRNAs | 89 |
| -ncRNAs | 5 |
| Organism | Genes | Core (%) | Accessory (%) | Unique (%) |
|---|---|---|---|---|
| Metabacillus sp. KIGAM252T | 3919 | 28.7 | 55.5 | 15.9 |
| Metabacillus crassostreae DSM 24486T | 4416 | 25.4 | 66.4 | 8.2 |
| Metabacillus fastidiosus NBRC 101226T | 4163 | 27.0 | 38.0 | 35.0 |
| Metabacillus halosaccharovorans DSM 25387T | 4786 | 23.5 | 58.0 | 18.5 |
| Metabacillus idriensis SMC4352-2T | 4599 | 24.4 | 54.3 | 21.3 |
| Metabacillus indicus LMG 22858T | 3704 | 30.3 | 59.2 | 10.5 |
| Metabacillus iocasae DSM 104297T | 3834 | 29.3 | 32.9 | 37.8 |
| Metabacillus lacus AK74T | 3767 | 29.8 | 41.1 | 29.1 |
| Metabacillus litoralis SW-211T | 4350 | 25.8 | 66.5 | 7.7 |
| Metabacillus mangrovi AK61T | 3947 | 28.5 | 55.6 | 15.9 |
| Metabacillus niabensis 4T19T | 4546 | 24.7 | 52.4 | 22.9 |
| Metabacillus sediminilitoris DSL-17T | 4670 | 24.0 | 51.4 | 24.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, C.Y.; Cho, E.-S.; Yoon, D.J.; Cha, I.-T.; Jung, D.-H.; Nam, Y.-D.; Park, S.-L.; Lim, S.-I.; Seo, M.-J. Genomic and Physiological Characterization of Metabacillus flavus sp. nov., a Novel Carotenoid-Producing Bacilli Isolated from Korean Marine Mud. Microorganisms 2022, 10, 979. https://doi.org/10.3390/microorganisms10050979
Hwang CY, Cho E-S, Yoon DJ, Cha I-T, Jung D-H, Nam Y-D, Park S-L, Lim S-I, Seo M-J. Genomic and Physiological Characterization of Metabacillus flavus sp. nov., a Novel Carotenoid-Producing Bacilli Isolated from Korean Marine Mud. Microorganisms. 2022; 10(5):979. https://doi.org/10.3390/microorganisms10050979
Chicago/Turabian StyleHwang, Chi Young, Eui-Sang Cho, Deok Jun Yoon, In-Tae Cha, Dong-Hyun Jung, Young-Do Nam, So-Lim Park, Seong-Il Lim, and Myung-Ji Seo. 2022. "Genomic and Physiological Characterization of Metabacillus flavus sp. nov., a Novel Carotenoid-Producing Bacilli Isolated from Korean Marine Mud" Microorganisms 10, no. 5: 979. https://doi.org/10.3390/microorganisms10050979
APA StyleHwang, C. Y., Cho, E.-S., Yoon, D. J., Cha, I.-T., Jung, D.-H., Nam, Y.-D., Park, S.-L., Lim, S.-I., & Seo, M.-J. (2022). Genomic and Physiological Characterization of Metabacillus flavus sp. nov., a Novel Carotenoid-Producing Bacilli Isolated from Korean Marine Mud. Microorganisms, 10(5), 979. https://doi.org/10.3390/microorganisms10050979