
Bacterial and Protistan Community Variation across the Changjiang Estuary to the Ocean with Multiple Environmental Gradients

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Environmental Parameters Measurements
2.3. DNA Extraction, PCR Amplification, and High-Throughput Sequencing
2.4. Sequence Processing
2.5. Statistical Analysis
2.6. Function Prediction
3. Results
3.1. Environmental Characteristics
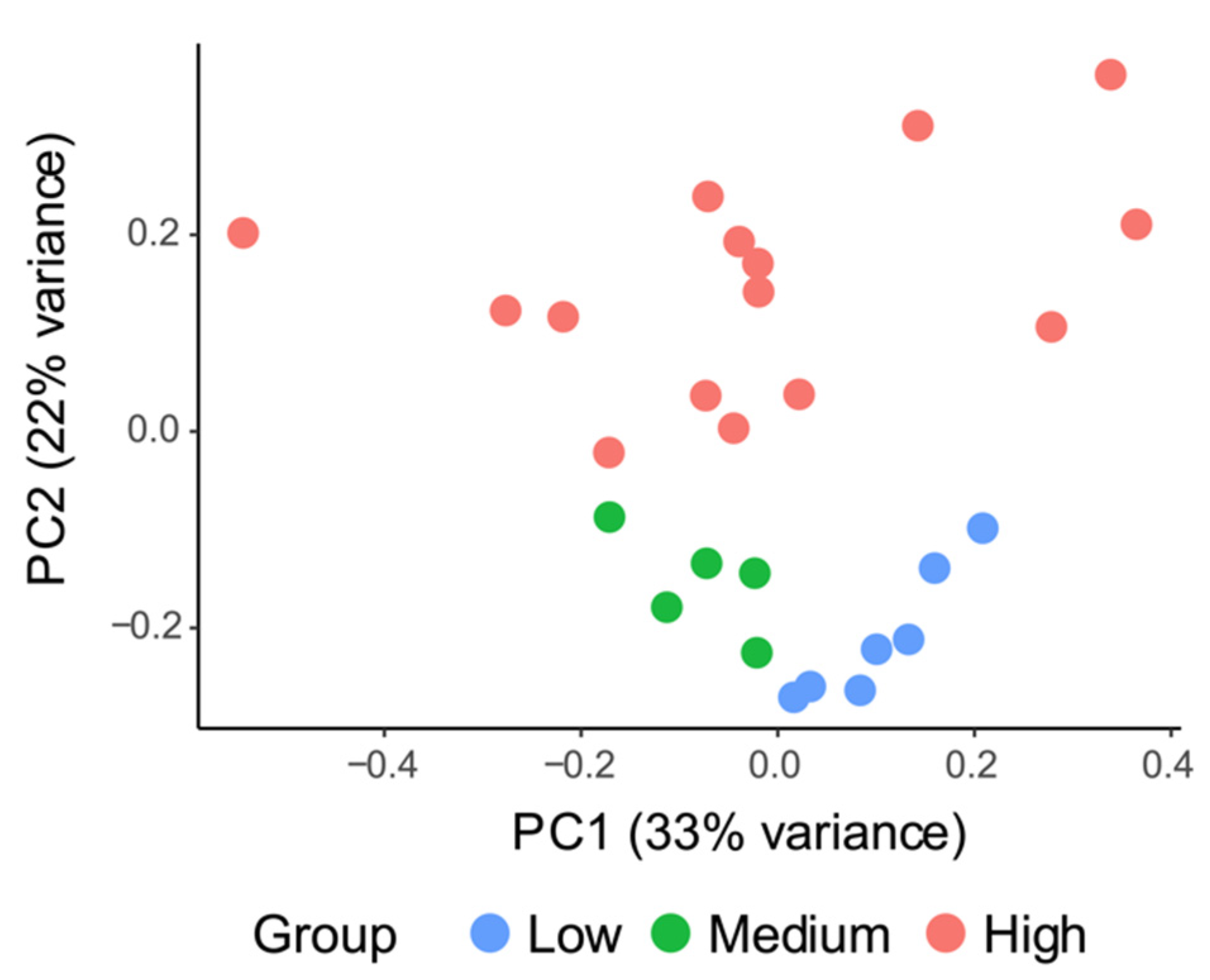
3.2. Spatial Patterns in Bacterial and Protistan Community
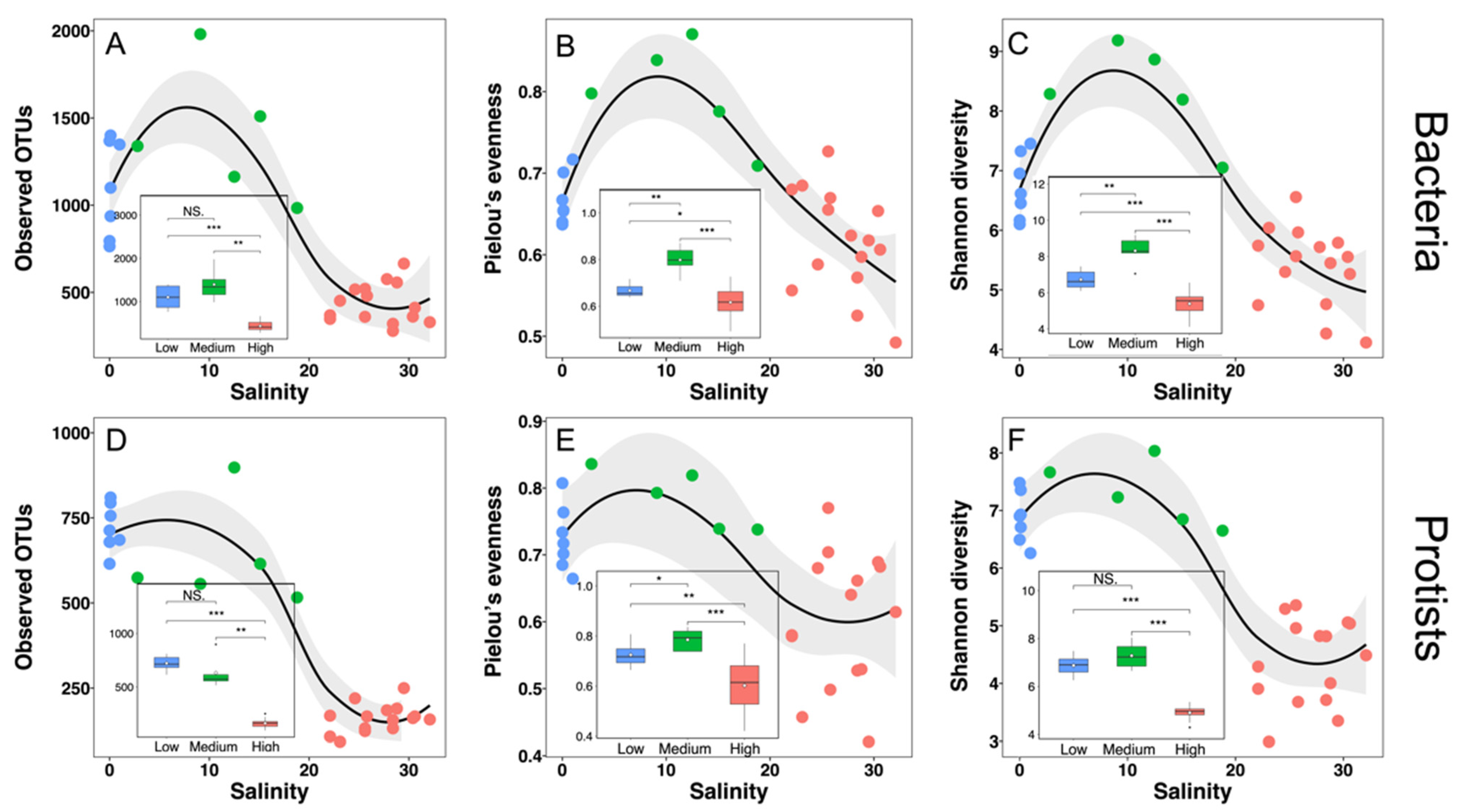
3.3. Alpha-Diversity Patterns
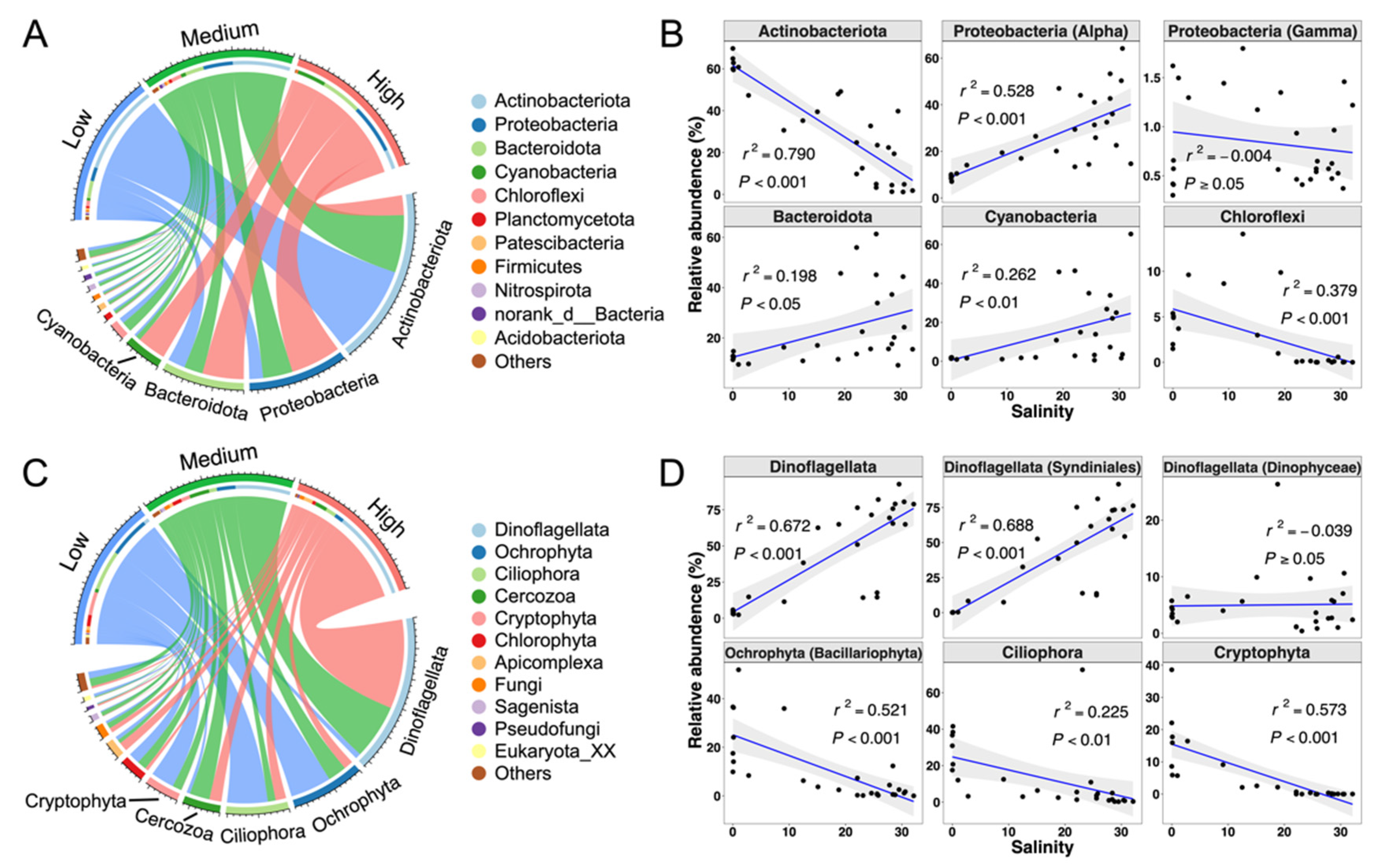
3.4. Community Composition
3.5. Profiles of Bacterial Functional Prediction
4. Discussion
4.1. Contribution of Salinity and Other Environmental Factors in Shaping Microbial Communities
4.2. Microbial Alpha Diversity and Microbial Community Composition Structured by Salinity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Green, J.L.; Bohannan, B.J.M.; Whitaker, R.J. Microbial biogeography: From taxonomy to traits. Science 2008, 320, 1039–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martiny, J.B.H.; Bohannan, B.J.M.; Brown, J.H.; Colwell, R.K.; Fuhrman, J.A.; Green, J.L.; Horner-Devine, M.C.; Kane, M.; Krumins, J.A.; Kuske, C.R.; et al. Microbial biogeography: Putting microorganisms on the map. Nat. Rev. Microbiol. 2006, 4, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Falkowski, P.G.; Fenchel, T.; Delong, E.F. The microbial engines that drive Earth’s biogeochemical cycles. Science 2008, 320, 1034–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozupone, C.A.; Knight, R. Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. USA 2007, 104, 11436–11440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Wu, L.; Huang, L. Spatiotemporal Patterns in Diversity and Assembly Process of Marine Protist Communities of the Changjiang (Yangtze River) Plume and Its Adjacent Waters. Front. Microbiol. 2020, 11, 579290. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.M.; Dai, Q.P.; Liu, X.Z.; Fan, Y.P.; Wang, J.X. Comparison of bacterial community structure and potential functions in hypoxic and non-hypoxic zones of the Changjiang Estuary. PLoS ONE 2019, 14, e0217431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Song, S.; Chen, T.; Li, C. The diversity and structure of marine protists in the coastal waters of China revealed by morphological observation and 454 pyrosequencing. Estuar. Coast. Shelf Sci. 2017, 189, 143–155. [Google Scholar] [CrossRef]
- Krause, E.; Wichels, A.; Giménez, L.; Lunau, M.; Schilhabel, M.B.; Gerdts, G. Small changes in pH have direct effects on marine bacterial community composition: A microcosm approach. PLoS ONE 2012, 7, e47035. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Juarez, D.L.; Pan, J.F.; Blinebry, S.K.; Gronniger, J.; Clark, J.S.; Johnson, Z.I.; Hunt, D.E. Microbial communities across nearshore to offshore coastal transects are primarily shaped by distance and temperature. Env. Microbiol. 2019, 21, 3862–3872. [Google Scholar] [CrossRef]
- Mason, O.U.; Canter, E.J.; Gillies, L.E.; Paisie, T.K.; Roberts, B.J. Mississippi River Plume Enriches Microbial Diversity in the Northern Gulf of Mexico. Front. Microbiol. 2016, 7, 1048. [Google Scholar] [CrossRef] [Green Version]
- Tee, H.S.; Waite, D.; Lear, G.; Handley, K.M. Microbial river-to-sea continuum: Gradients in benthic and planktonic diversity, osmoregulation and nutrient cycling. Microbiome 2021, 9, 190. [Google Scholar] [CrossRef]
- Fortunato, C.S.; Crump, B.C. Bacterioplankton Community Variation Across River to Ocean Environmental Gradients. Microb. Ecol. 2011, 62, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, C.S.; Crump, B.C. Microbial gene abundance and expression patterns across a river to ocean salinity gradient. PLoS ONE 2015, 10, e0140578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logares, R.; Tesson, S.V.; Canbäck, B.; Pontarp, M.; Hedlund, K.; Rengefors, K. Contrasting prevalence of selection and drift in the community structuring of bacteria and microbial eukaryotes. Environ. Microbiol. 2018, 20, 2231–2240. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Song, X.; Zhang, C.Y.; Chen, G.F.; Lao, Y.M.; Jin, H.; Cai, Z.H. Distribution Patterns of Microbial Community Structure Along a 7000-Mile Latitudinal Transect from the Mediterranean Sea Across the Atlantic Ocean to the Brazilian Coastal Sea. Microb. Ecol. 2018, 76, 592–609. [Google Scholar] [CrossRef]
- Gao, L.; Li, D.; Ishizaka, J.; Zhang, Y.; Zong, H.; Guo, L. Nutrient dynamics across the river-sea interface in the Changjiang (Yangtze River) estuary-East China Sea region. Limnol. Oceanogr. 2015, 60, 2207–2221. [Google Scholar] [CrossRef] [Green Version]
- Damashek, J.; Casciotti, K.L.; Francis, C.A. Variable Nitrification Rates Across Environmental Gradients in Turbid, Nutrient-Rich Estuary Waters of San Francisco Bay. Estuaries Coasts 2016, 39, 1050–1071. [Google Scholar] [CrossRef]
- Remane, A. Die Brackwasserfauna: Mit besonderer Berücksichtigung der Ostsee. Zool. Anz. 1934, 7, 34–74. [Google Scholar]
- Herlemann, D.P.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- Filker, S.; Kuhner, S.; Heckwolf, M.; Dierking, J.; Stoeck, T. A fundamental difference between macrobiota and microbial eukaryotes: Protistan plankton has a species maximum in the freshwater-marine transition zone of the Baltic Sea. Environ. Microbiol. 2019, 21, 603–617. [Google Scholar] [CrossRef]
- Hu, Y.O.; Karlson, B.; Charvet, S.; Andersson, A.F. Diversity of Pico- to Mesoplankton along the 2000 km Salinity Gradient of the Baltic Sea. Front. Microbiol. 2016, 7, 679. [Google Scholar] [CrossRef] [PubMed]
- Romero, O.E.; Armand, L.K. Marine diatoms as indicators of modern changes in oceanographic conditions. In The Diatoms: Applications for the Environmental and Earth Sciences, Second Edition; Cambridge University Press: Cambridge, UK, 2010; pp. 373–400. [Google Scholar]
- Burliga, A.L.; Kociolek, J.P. Diatoms (Bacillariophyta) in rivers. In River Algae; Springer: Berlin/Heidelberg, Germany, 2016; pp. 93–128. [Google Scholar]
- Wu, Z.; Cai, Y.; Liu, X.; Xu, C.P.; Chen, Y.; Zhang, L. Temporal and spatial variability of phytoplankton in Lake Poyang: The largest freshwater lake in China. J. Great Lakes Res. 2013, 39, 476–483. [Google Scholar] [CrossRef]
- Li, W.K. Primary production of prochlorophytes, cyanobacteria, and eucaryotic ultraphytoplankton: Measurements from flow cytometric sorting. Limnol. Oceanogr. 1994, 39, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Song, J. A numerical study of the seasonal variations of nutrients in the Changjiang River estuary and its adjacent sea area. Ecol. Model. 2014, 291, 69–81. [Google Scholar] [CrossRef]
- Li, M.; Xu, K.; Watanabe, M.; Chen, Z. Long-term variations in dissolved silicate, nitrogen, and phosphorus flux from the Yangtze River into the East China Sea and impacts on estuarine ecosystem. Estuar. Coast. Shelf Sci. 2007, 71, 3–12. [Google Scholar] [CrossRef]
- Zhiliang, S.; Qun, L.; Shumei, Z.; Hui, M.; Ping, Z. A nitrogen budget of the Changjiang River catchment. AMBIO J. Hum. Environ. 2003, 32, 65–69. [Google Scholar] [CrossRef]
- Yan, W.; Zhang, S.; Sun, P.; Seitzinger, S.P. How do nitrogen inputs to the Changjiang basin impact the Changjiang River nitrate: A temporal analysis for 1968–1997. Glob. Biogeochem. Cycles 2003, 17. [Google Scholar] [CrossRef] [Green Version]
- Tong, Y.; Zhao, Y.; Zhen, G.; Chi, J.; Liu, X.; Lu, Y.; Wang, X.; Yao, R.; Chen, J.; Zhang, W. Nutrient loads flowing into coastal waters from the main rivers of China (2006–2012). Sci. Rep. 2015, 5, 16678. [Google Scholar] [CrossRef]
- Liu, X.; Shen, H. Estimation of dissolved inorganic nutrients fluxes from the Changjiang River into estuary. Sci. China Ser. B Chem. 2001, 44, 135–141. [Google Scholar] [CrossRef]
- Feng, B.W.; Li, X.R.; Wang, J.H.; Hu, Z.Y.; Meng, H.; Xiang, L.Y.; Quan, Z.X. Bacterial diversity of water and sediment in the Changjiang estuary and coastal area of the East China Sea. FEMS Microbiol. Ecol. 2009, 70, 80–92. [Google Scholar] [CrossRef]
- Guo, W.; Yang, L.; Zhai, W.; Chen, W.; Osburn, C.L.; Huang, X.; Li, Y. Runoff-mediated seasonal oscillation in the dynamics of dissolved organic matter in different branches of a large bifurcated estuary—The Changjiang Estuary. J. Geophys. Res. Biogeosci. 2014, 119, 776–793. [Google Scholar] [CrossRef]
- Ning, X.; Lin, C.; Su, J.; Liu, C.; Hao, Q.; Le, F. Long-term changes of dissolved oxygen, hypoxia, and the responses of the ecosystems in the East China Sea from 1975 to 1995. J. Oceanogr. 2011, 67, 59–75. [Google Scholar] [CrossRef]
- Jiang, Z.; Chen, J.; Zhou, F.; Shou, L.; Chen, Q.; Tao, B.; Yan, X.; Wang, K. Controlling factors of summer phytoplankton community in the Changjiang (Yangtze River) Estuary and adjacent East China Sea shelf. Cont. Shelf Res. 2015, 101, 71–84. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, P.; Lian, E.; Wu, H.; Liu, D. Spatial distribution of chlorophyll a and its relationships with environmental factors influenced by front in the Changjiang River Estuary and its adjacent waters in summer 2019. Mar. Sci. Bull. 2021, 40, 541–549. [Google Scholar]
- Stoeck, T.; Bass, D.; Nebel, M.; Christen, R.; Jones, M.D.; Breiner, H.W.; Richards, T.A. Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol. Ecol. 2010, 19 (Suppl. S1), 21–31. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet 2011, 17, 10. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Pielou, E.C. The measurement of diversity in different types of biological collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2′s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Schlitzer, R. Ocean Data View. Available online: https://odv.awi.de/ (accessed on 2 June 2020).
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Stevens, M.H.H.; Oksanen, M.J.; Suggests, M. The vegan package. Community Ecol. Package 2007, 10, 719. [Google Scholar]
- Harrell, F.E., Jr.; Harrell, M.F.E., Jr. Package ‘hmisc’. CRAN2018 2019, 2019, 235–236. [Google Scholar]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 326–349. [Google Scholar] [CrossRef]
- Ramette, A. Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol. 2007, 62, 142–160. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.; Zou, Y.; Zhang, J.; Peres-Neto, P. Rdacca.hp: An R package for generalizing hierarchical and variation partitioning in multiple regression and canonical analysis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Caicedo, H.H.; Hashimoto, D.A.; Caicedo, J.C.; Pentland, A.; Pisano, G.P. Overcoming barriers to early disease intervention. Nat. Biotechnol. 2020, 38, 669–673. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Liu, Y.X.; Huang, L. ImageGP: An easy-to-use data visualization web server for scientific researchers. iMeta 2022, 1, e5. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Krieger, A.M.; Yekutieli, D. Adaptive linear step-up procedures that control the false discovery rate. Biometrika 2006, 93, 491–507. [Google Scholar] [CrossRef]
- Thompson, L.R.; Williams, G.J.; Haroon, M.F.; Shibl, A.; Larsen, P.; Shorenstein, J.; Knight, R.; Stingl, U. Metagenomic covariation along densely sampled environmental gradients in the Red Sea. ISME J. 2017, 11, 138–151. [Google Scholar] [CrossRef] [Green Version]
- Doherty, M.; Yager, P.L.; Moran, M.A.; Coles, V.J.; Fortunato, C.S.; Krusche, A.V.; Medeiros, P.M.; Payet, J.P.; Richey, J.E.; Satinsky, B.M.; et al. Bacterial Biogeography across the Amazon River-Ocean Continuum. Front. Microbiol. 2017, 8, 882. [Google Scholar] [CrossRef]
- Witkowski, A.; Broszinski, A.; Bennike, O.; Janczak-Kostecka, B.; Jensen, J.B.; Lemke, W.; Endler, R.; Kuijpers, A. Darss Sill as a biological border in the fossil record of the Baltic Sea: Evidence from diatoms. Quat. Int. 2005, 130, 97–109. [Google Scholar] [CrossRef]
- Steele, J.A.; Countway, P.D.; Xia, L.; Vigil, P.D.; Beman, J.M.; Kim, D.Y.; Chow, C.-E.T.; Sachdeva, R.; Jones, A.C.; Schwalbach, M.S.; et al. Marine bacterial, archaeal and protistan association networks reveal ecological linkages. Isme J. 2011, 5, 1414–1425. [Google Scholar] [CrossRef]
- Stoecker, D.K. Mixotrophy among Dinoflagellates1. J. Eukaryot. Microbiol. 1999, 46, 397–401. [Google Scholar] [CrossRef]
- Weisse, T. Functional diversity of aquatic ciliates. Eur. J. Protistol. 2017, 61, 331–358. [Google Scholar] [CrossRef]
- Guillou, L.; Viprey, M.; Chambouvet, A.; Welsh, R.M.; Kirkham, A.R.; Massana, R.; Scanlan, D.J.; Worden, A.Z. Widespread occurrence and genetic diversity of marine parasitoids belonging to Syndiniales (Alveolata). Env. Microbiol. 2008, 10, 3349–3365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jephcott, T.G.; Alves-de-Souza, C.; Gleason, F.H.; van Ogtrop, F.F.; Sime-Ngando, T.; Karpov, S.A.; Guillou, L. Ecological impacts of parasitic chytrids, syndiniales and perkinsids on populations of marine photosynthetic dinoflagellates. Fungal Ecol. 2016, 19, 47–58. [Google Scholar] [CrossRef]
- Fortunato, C.S.; Herfort, L.; Zuber, P.; Baptista, A.M.; Crump, B.C. Spatial variability overwhelms seasonal patterns in bacterioplankton communities across a river to ocean gradient. Isme J. 2012, 6, 554–563. [Google Scholar] [CrossRef]
- Telesh, I.V.; Schubert, H.; Skarlato, S.O. Revisiting Remane’s concept: Evidence for high plankton diversity and a protistan species maximum in the horohalinicum of the Baltic Sea. Mar. Ecol. Prog. 2011, 421, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Oren, A. Life at high salt concentrations. Prokaryotes 2006, 3, 263–282. [Google Scholar]
- Wang, J.; Yang, D.; Zhang, Y.; Shen, J.; van der Gast, C.; Hahn, M.W.; Wu, Q. Do Patterns of Bacterial Diversity along Salinity Gradients Differ from Those Observed for Macroorganisms? PLoS ONE 2011, 6, e27597. [Google Scholar] [CrossRef] [Green Version]
- Herlemann, D.P.R.; Lundin, D.; Andersson, A.F.; Labrenz, M.; Juergens, K. Phylogenetic Signals of Salinity and Season in Bacterial Community Composition Across the Salinity Gradient of the Baltic Sea. Front. Microbiol. 2016, 7, 1883. [Google Scholar] [CrossRef]
- Snoeijs-Leijonmalm, P.; Andrén, E. Why is the Baltic Sea so special to live in? In Biological Oceanography of the Baltic Sea; Snoeijs-Leijonmalm, H.S., Radziejewska, T., Eds.; Springer: Dordrecht, The Netherlands, 2017; pp. 23–84. [Google Scholar]
- Pavloudi, C.; Kristoffersen, J.B.; Oulas, A.; De Troch, M.; Arvanitidis, C. Sediment microbial taxonomic and functional diversity in a natural salinity gradient challenge Remane’s “species minimum” concept. Peerj 2017, 5, e3687. [Google Scholar] [CrossRef] [Green Version]
- Crump, B.C.; Armbrust, E.V.; Baross, J.A. Phylogenetic analysis of particle-attached and free-living bacterial communities in the Columbia river, its estuary, and the adjacent coastal ocean. Appl. Environ. Microbiol. 1999, 65, 3192–3204. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.W. The intermediate disturbance hypothesis should be abandoned. Trends Ecol. Evol. 2013, 28, 86–92. [Google Scholar] [CrossRef]
- Filker, S.; Forster, D.; Weinisch, L.; Mora-Ruiz, M.; González, B.; Farías, M.E.; Rosselló-Móra, R.; Stoeck, T. Transition boundaries for protistan species turnover in hypersaline waters of different biogeographic regions. Environ. Microbiol. 2017, 19, 3186–3200. [Google Scholar] [CrossRef] [PubMed]
- Forster, D.; Behnke, A.; Stoeck, T. Meta-analyses of environmental sequence data identify anoxia and salinity as parameters shaping ciliate communities. Syst. Biodivers. 2012, 10, 277–288. [Google Scholar] [CrossRef]
- Campbell, B.J.; Kirchman, D.L. Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient. Isme J. 2013, 7, 210–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Zhang, H.; Liu, P.; Wang, Y.; Sun, Y.; Song, Z.; Hu, X. Divergent Patterns of Bacterial Community Structure and Function in Response to Estuarine Output in the Middle of the Bohai Sea. Front. Microbiol. 2021, 12, 630741. [Google Scholar] [CrossRef]
- Mehrshad, M.; Salcher, M.M.; Okazaki, Y.; Nakano, S.-I.; Šimek, K.; Andrei, A.-S.; Ghai, R. Hidden in plain sight—highly abundant and diverse planktonic freshwater Chloroflexi. Microbiome 2018, 6, 176. [Google Scholar] [CrossRef]
- Elloumi, J.; Carrias, J.-F.; Ayadi, H.; Sime-Ngando, T.; Boukhris, M.; Bouaïn, A. Composition and distribution of planktonic ciliates from ponds of different salinity in the solar saltwork of Sfax, Tunisia. Estuar. Coast. Shelf Sci. 2006, 67, 21–29. [Google Scholar] [CrossRef]
- Piwosz, K.; Całkiewicz, J.; Gołębiewski, M.; Creer, S. Diversity and community composition of pico-and nanoplanktonic protists in the Vistula River estuary (Gulf of Gdańsk, Baltic Sea). Estuar. Coast. Shelf Sci. 2018, 207, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Liu, J.; Chen, J.; Chen, Q.; Yan, X.; Xuan, J.; Zeng, J. Responses of summer phytoplankton community to drastic environmental changes in the Changjiang (Yangtze River) estuary during the past 50 years. Water Res. 2014, 54, 1–11. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, J.; Zheng, B.; Cai, W.; Lin, K.; Tang, J. Temporal and spatial distribution of red tide outbreaks in the Yangtze River Estuary and adjacent waters, China. Mar. Pollut. Bull. 2013, 72, 213–221. [Google Scholar] [CrossRef]
- Balzano, S.; Abs, E.; Leterme, S.C. Protist diversity along a salinity gradient in a coastal lagoon. Aquat. Microb. Ecol. 2015, 74, 263–277. [Google Scholar]
- Moon-van der Staay, S.Y.; De Wachter, R.; Vaulot, D. Oceanic 18S rDNA sequences from picoplankton reveal unsuspected eukaryotic diversity. Nature 2001, 409, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Countway, P.D.; Gast, R.J.; Savai, P.; Caron, D.A. Protistan diversity estimates based on 18S rDNA from seawater incubations in the western North Atlantic 1. J. Eukaryot. Microbiol. 2005, 52, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Massana, R.; Not, F.; Marie, D.; Vaulot, D. Mapping of picoeucaryotes in marine ecosystems with quantitative PCR of the 18S rRNA gene. FEMS Microbiol. Ecol. 2005, 52, 79–92. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Dong, J.; Liu, X.; Massana, R. Extremely high copy numbers and polymorphisms of the rDNA operon estimated from single cell analysis of oligotrich and peritrich ciliates. Protist 2013, 164, 369–379. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Simple Mantel Test | Partial Mantel Test | ||||
|---|---|---|---|---|---|
| Bacteria | Protists | Bacteria | Protists | ||
| Effects of | r | r | Control for | r | r |
| Salinity | 0.884 *** | 0.864 *** | DIN | 0.421 *** | 0.496 *** |
| DIP | 0.875 *** | 0.857 *** | |||
| DSi | 0.497 *** | 0.517 *** | |||
| Geo | 0.778 *** | 0.819 *** | |||
| DIN | 0.866 *** | 0.816 *** | Salinity | 0.249 *** | 0.056 |
| DIP | 0.859 *** | 0.811 *** | |||
| DSi | 0.308 *** | 0.205 ** | |||
| Geo | 0.754 *** | 0.740 *** | |||
| DIP | 0.263 ** | 0.226 ** | Salinity | 0.048 | −0.02 |
| DIN | −0.15 | −0.162 | |||
| DSi | −0.077 | −0.11 | |||
| Geo | 0.208 ** | 0.167 * | |||
| DSi | 0.854 *** | 0.814 *** | Salinity | 0.267 *** | 0.139 * |
| DIN | 0.144 * | 0.174 * | |||
| DIP | 0.843 *** | 0.805 *** | |||
| Geo | 0.719 *** | 0.740 *** | |||
| Geo | 0.707 *** | 0.513 *** | Salinity | 0.313 *** | −0.198 |
| DIN | 0.370 *** | −0.049 | |||
| DIP | 0.697 *** | 0.495 *** | |||
| DSi | 0.328 *** | −0.098 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, X.; Zhu, Z.; Wu, J.; Lian, E.; Liu, D.; Yang, S.; Zhang, R. Bacterial and Protistan Community Variation across the Changjiang Estuary to the Ocean with Multiple Environmental Gradients. Microorganisms 2022, 10, 991. https://doi.org/10.3390/microorganisms10050991
Jiang X, Zhu Z, Wu J, Lian E, Liu D, Yang S, Zhang R. Bacterial and Protistan Community Variation across the Changjiang Estuary to the Ocean with Multiple Environmental Gradients. Microorganisms. 2022; 10(5):991. https://doi.org/10.3390/microorganisms10050991
Chicago/Turabian StyleJiang, Xinjun, Zhu Zhu, Jinnan Wu, Ergang Lian, Dongyan Liu, Shouye Yang, and Ruifeng Zhang. 2022. "Bacterial and Protistan Community Variation across the Changjiang Estuary to the Ocean with Multiple Environmental Gradients" Microorganisms 10, no. 5: 991. https://doi.org/10.3390/microorganisms10050991
APA StyleJiang, X., Zhu, Z., Wu, J., Lian, E., Liu, D., Yang, S., & Zhang, R. (2022). Bacterial and Protistan Community Variation across the Changjiang Estuary to the Ocean with Multiple Environmental Gradients. Microorganisms, 10(5), 991. https://doi.org/10.3390/microorganisms10050991