
An In Silico Analysis of Malaria Pre-Erythrocytic-Stage Antigens Interpreting Worldwide Genetic Data to Suggest Vaccine Candidate Variants and Epitopes

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antigen Selection
2.2. Allelic Sequence Generation
2.3. Data Analyses
2.3.1. Nucleotide Polymorphism
2.3.2. Haplotype Diversity and Distribution
2.3.3. Population Structure
2.3.4. Cytotoxic T-Lymphocyte (CTL) Epitope Identification and Polymorphisms
2.3.5. B-Cell Epitopes and Polymorphisms
2.3.6. Polymorphisms in Epitopes and Protein Function
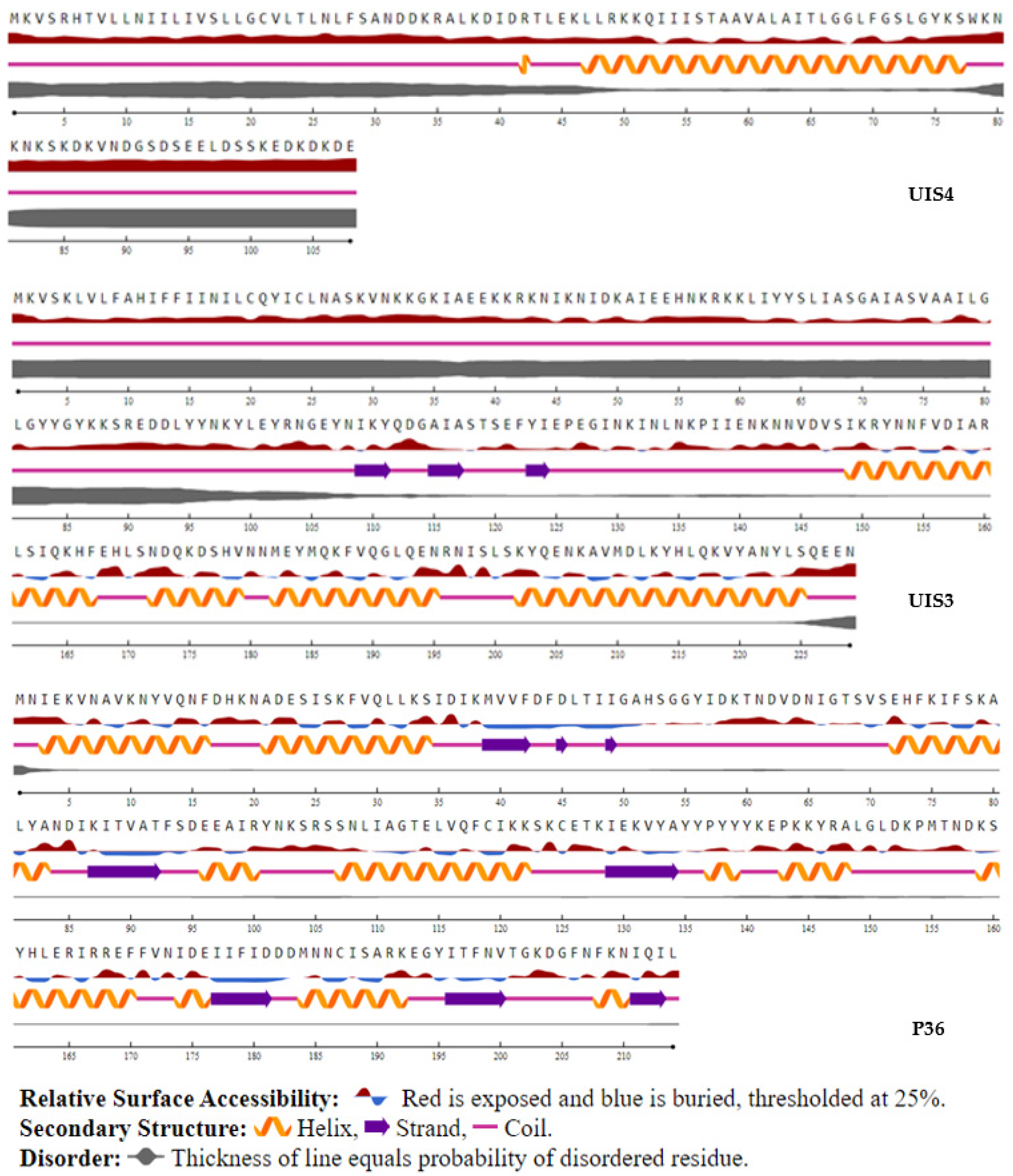
2.3.7. Secondary Predicted Structure and Solvent Accessibility
3. Results
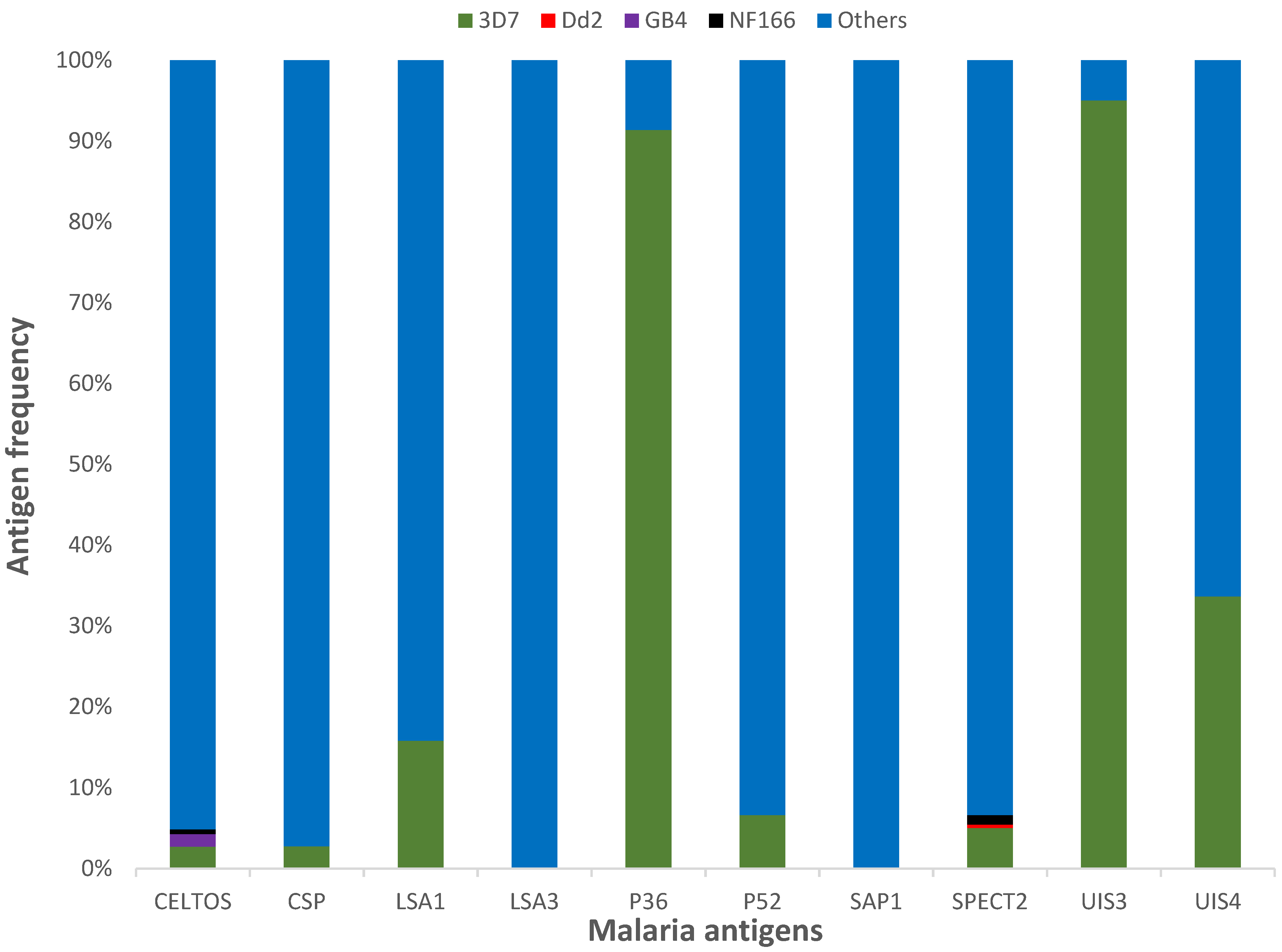
3.1. Protein Diversity and Mode of Evolution
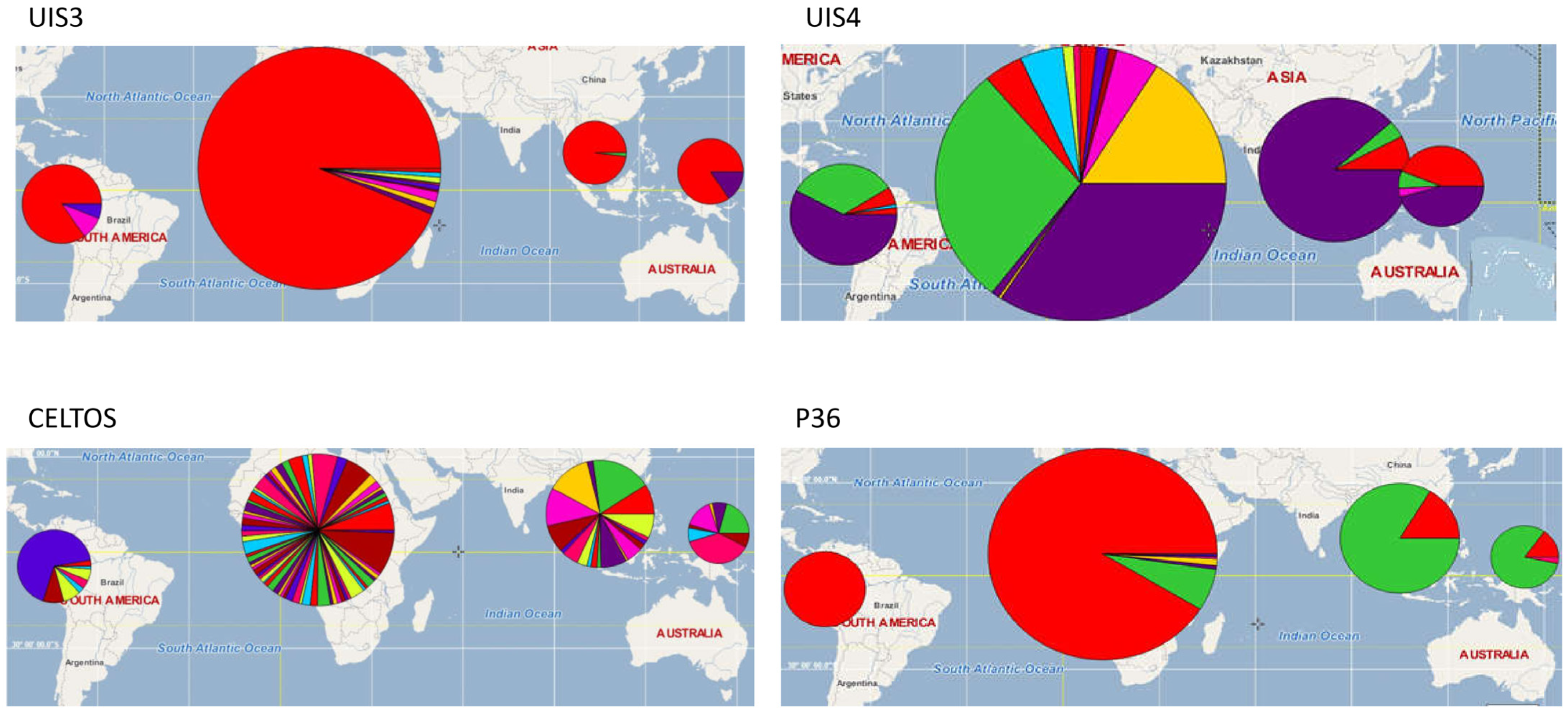
3.2. Vaccine Antigen Geographic Distribution and Haplotype Diversity
3.3. Identification of T-Cell Epitope and Polymorphisms
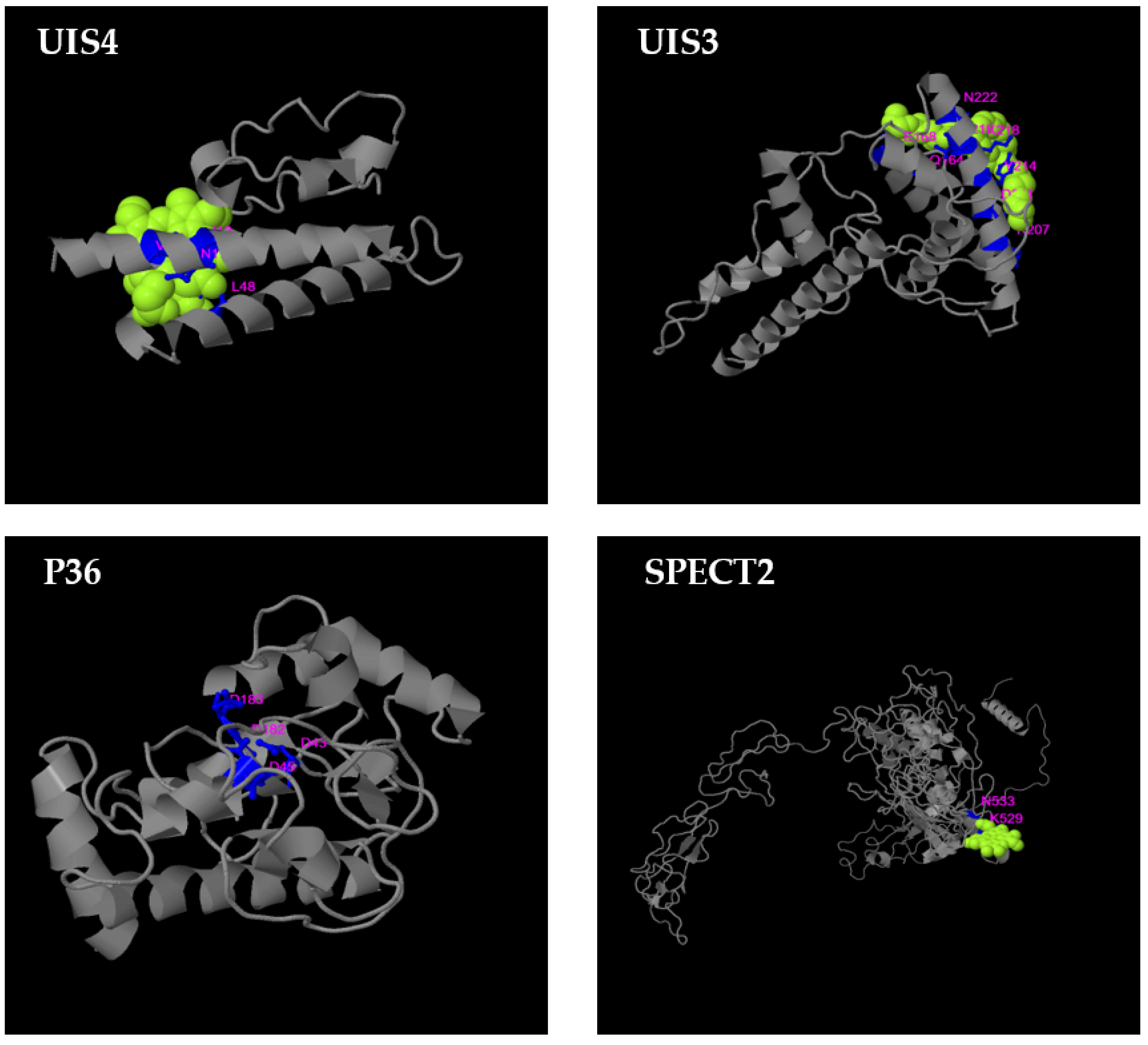
3.4. B-Cell Conformational Epitopes and Polymorphisms
3.5. T-Cell Epitope Biological Relevance and Solvent Accessibility
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tran, T.M.; Portugal, S.; Draper, S.J.; Crompton, P.D. Malaria Vaccines: Moving Forward After Encouraging First Steps. Curr. Trop. Med. Rep. 2015, 2, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, M.; de Savigny, D. Malaria eradication back on the table. Bull. World Health Organ. 2008, 86, 82. [Google Scholar] [CrossRef] [PubMed]
- mal ERACGoV. A research agenda for malaria eradication: Vaccines. PLoS Med. 2011, 8, e1000398. [Google Scholar] [CrossRef]
- Henderson, D.A. The miracle of vaccination. Notes Rec. R. Soc. Lond. 1997, 51, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Delany, I.; Rappuoli, R.; De, G.E. Vaccines for the 21st century. EMBO Mol. Med. 2014, 6, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Genton, B.; Al-Yaman, F.; Betuela, I.; Anders, R.F.; Saul, A.; Baea, K.; Mellombo, M.; Taraika, J.; Brown, G.V.; Pye, D.; et al. Safety and immunogenicity of a three-component blood-stage malaria vaccine (MSP1, MSP2, RESA) against Plasmodium falciparum in Papua New Guinean children. Vaccine 2003, 22, 30–41. [Google Scholar] [CrossRef]
- Thera, M.A.; Doumbo, O.K.; Coulibaly, D.; Laurens, M.B.; Ouattara, A.; Kone, A.K.; Guindo, A.B.; Traore, K.; Traore, I.; Kouriba, B.; et al. A field trial to assess a blood-stage malaria vaccine. N. Engl. J. Med. 2011, 365, 1004–1013. [Google Scholar] [CrossRef] [Green Version]
- Ouattara, A.; Takala-Harrison, S.; Thera, M.A.; Coulibaly, D.; Niangaly, A.; Saye, R.; Tolo, Y.; Dutta, S.; Heppner, D.G.; Soisson, L.; et al. Molecular basis of allele-specific efficacy of a blood-stage malaria vaccine: Vaccine development implications. J. Infect. Dis. 2013, 207, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Neafsey, D.E.; Juraska, M.; Bedford, T.; Benkeser, D.; Valim, C.; Griggs, A.; Lievens, M.; Abdulla, S.; Adjei, S.; Agbenyega, T.; et al. Genetic Diversity and Protective Efficacy of the RTS,S/AS01 Malaria Vaccine. N. Engl. J. Med. 2015, 373, 2025–2037. [Google Scholar] [CrossRef]
- Obaro, S.K. Confronting the pneumococcus: A target shift or bullet change? Vaccine 2000, 19, 1211–1217. [Google Scholar] [CrossRef]
- Obaro, S.K. The new pneumococcal vaccine. Clin. Microbiol. Infect. 2002, 8, 623–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diggle, M.A.; Clarke, S.C. Increased genetic diversity of Neisseria meningitidis isolates after the introduction of meningococcal serogroup C polysaccharide conjugate vaccines. J. Clin. Microbiol. 2005, 43, 4649–4653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolland, M.; Tovanabutra, S.; de Camp, A.C.; Frahm, N.; Gilbert, P.B.; Sanders-Buell, E.; Heath, L.; Magaret, C.A.; Bose, M.; Bradfield, A.; et al. Genetic impact of vaccination on breakthrough HIV-1 sequences from the STEP trial. Nat. Med. 2011, 17, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Edlefsen, P.T.; Rolland, M.; Hertz, T.; Tovanabutra, S.; Gartland, A.J.; de Camp, A.C.; Magaret, C.A.; Ahmed, H.; Gottardo, R.; Juraska, M.; et al. Comprehensive sieve analysis of breakthrough HIV-1 sequences in the RV144 vaccine efficacy trial. PLoS Comput. Biol. 2015, 11, e1003973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, A.E.; Schultz, L.; Buckee, C.O.; Reeder, J.C. Contrasting population structures of the genes encoding ten leading vaccine-candidate antigens of the human malaria parasite, Plasmodium falciparum. PLoS ONE 2009, 4, e8497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, K.; Thera, M.A.; Coulibaly, D.; Traore, K.; Guindo, A.B.; Ouattara, A.; Takala-Harrison, S.; Berry, A.A.; Doumbo, O.K.; Plowe, C.V. Variation in the circumsporozoite protein of Plasmodium falciparum: Vaccine development implications. PLoS ONE 2014, 9, e101783. [Google Scholar] [CrossRef] [PubMed]
- Barry, A.E.; Schultz, L.; Senn, N.; Nale, J.; Kiniboro, B.; Siba, P.M.; Mueller, I.; Reeder, J.C. High levels of genetic diversity of Plasmodium falciparum populations in Papua New Guinea despite variable infection prevalence. Am. J. Trop. Med. Hyg. 2013, 88, 718–725. [Google Scholar] [CrossRef]
- Amambua-Ngwa, A.; Tetteh, K.K.; Manske, M.; Gomez-Escobar, N.; Stewart, L.B.; Deerhake, M.E.; Cheeseman, I.H.; Newbold, C.I.; Holder, A.A.; Knuepfer, E.; et al. Population genomic scan for candidate signatures of balancing selection to guide antigen characterization in malaria parasites. PLoS Genet. 2012, 8, e1002992. [Google Scholar] [CrossRef] [Green Version]
- Mobegi, V.A.; Duffy, C.W.; Amambua-Ngwa, A.; Loua, K.M.; Laman, E.; Nwakanma, D.C.; MacInnis, B.; Aspeling-Jones, H.; Murray, L.; Clark, T.G.; et al. Genome-wide analysis of selection on the malaria parasite Plasmodium falciparum in West African populations of differing infection endemicity. Mol. Biol. Evol. 2014, 31, 1490–1499. [Google Scholar] [CrossRef] [Green Version]
- Duffy, C.W.; Amambua-Ngwa, A.; Ahouidi, A.D.; Diakite, M.; Awandare, G.A.; Ba, H.; Tarr, S.J.; Murray, L.; Stewart, L.B.; D’Alessandro, U.; et al. Multi-population genomic analysis of malaria parasites indicates local selection and differentiation at the gdv1 locus regulating sexual development. Sci. Rep. 2018, 8, 15763. [Google Scholar] [CrossRef]
- Shelton, J.M.; Corran, P.; Risley, P.; Silva, N.; Hubbart, C.; Jeffreys, A.; Rowlands, K.; Craik, R.; Cornelius, V.; Hensmann, M.; et al. Genetic determinants of anti-malarial acquired immunity in a large multi-centre study. Malar. J. 2015, 14, 333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann-Leitner, E.S.; Mease, R.M.; De La Vega, P.; Savranskaya, T.; Polhemus, M.; Ockenhouse, C.; Angov, E. Immunization with pre-erythrocytic antigen CelTOS from Plasmodium falciparum elicits cross-species protection against heterologous challenge with Plasmodium berghei. PLoS ONE 2010, 5, e12294. [Google Scholar] [CrossRef] [Green Version]
- John, C.C.; Moormann, A.M.; Pregibon, D.C.; Sumba, P.O.; McHugh, M.M.; Narum, D.L.; Lanar, D.E.; Schluchter, M.D.; Kazura, J.W. Correlation of high levels of antibodies to multiple pre-erythrocytic Plasmodium falciparum antigens and protection from infection. Am. J. Trop. Med. Hyg. 2005, 73, 222–228. [Google Scholar] [CrossRef] [PubMed]
- John, C.C.; Tande, A.J.; Moormann, A.M.; Sumba, P.O.; Lanar, D.E.; Min, X.M.; Kazura, J.W. Antibodies to pre-erythrocytic Plasmodium falciparum antigens and risk of clinical malaria in Kenyan children. J. Infect. Dis. 2008, 197, 519–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, G.; Prieur, E.; Roussilhon, C.; Druilhe, P. Pre-Clinical Assessment of Novel Multivalent MSP3 Malaria Vaccine Constructs. PLoS ONE 2011, 6, e28165. [Google Scholar] [CrossRef]
- van Dijk, M.R.; Douradinha, B.; Franke-Fayard, B.; Heussler, V.; van Dooren, M.W.; van, S.B.; van Gemert, G.J.; Sauerwein, R.W.; Mota, M.M.; Waters, A.P.; et al. Genetically attenuated, P36p-deficient malarial sporozoites induce protective immunity and apoptosis of infected liver cells. Proc. Natl. Acad. Sci. USA 2005, 102, 12194–12199. [Google Scholar] [CrossRef] [Green Version]
- Van Buskirk, K.M.; O’Neill, M.T.; De La, V.; Maier, A.G.; Krzych, U.; Williams, J.; Dowler, M.G.; Sacci, J.B., Jr.; Kangwanrangsan, N.; Tsuboi, T.; et al. Preerythrocytic, live-attenuated Plasmodium falciparum vaccine candidates by design. Proc. Natl. Acad. Sci. USA 2009, 106, 13004–13009. [Google Scholar] [CrossRef] [Green Version]
- Mueller, A.K.; Labaied, M.; Kappe, S.H.; Matuschewski, K. Genetically modified Plasmodium parasites as a protective experimental malaria vaccine. Nature 2005, 433, 164–167. [Google Scholar] [CrossRef]
- Mueller, A.K.; Camargo, N.; Kaiser, K.; Andorfer, C.; Frevert, U.; Matuschewski, K.; Kappe, S.H. Plasmodium liver stage developmental arrest by depletion of a protein at the parasite-host interface. Proc. Natl. Acad. Sci. USA 2005, 102, 3022–3027. [Google Scholar] [CrossRef] [Green Version]
- Aly, A.S.; Mikolajczak, S.A.; Rivera, H.S.; Camargo, N.; Jacobs-Lorena, V.; Labaied, M.; Coppens, I.; Kappe, S.H. Targeted deletion of SAP1 abolishes the expression of infectivity factors necessary for successful malaria parasite liver infection. Mol. Microbiol. 2008, 69, 152–163. [Google Scholar] [CrossRef] [Green Version]
- Kappe, S.H.; Buscaglia, C.A.; Bergman, L.W.; Coppens, I.; Nussenzweig, V. Apicomplexan gliding motility and host cell invasion: Overhauling the motor model. Trends Parasitol. 2004, 20, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Takala, S.L.; Escalante, A.A.; Branch, O.H.; Kariuki, S.; Biswas, S.; Chaiyaroj, S.C.; Lal, A.A. Genetic diversity in the Block 2 region of the merozoite surface protein 1 (MSP-1) of Plasmodium falciparum: Additional complexity and selection and convergence in fragment size polymorphism. Infect. Genet. Evol. 2006, 6, 417–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takala, S.L.; Coulibaly, D.; Thera, M.A.; Batchelor, A.H.; Cummings, M.P.; Escalante, A.A.; Ouattara, A.; Traore, K.; Niangaly, A.; Djimde, A.A.; et al. Extreme polymorphism in a vaccine antigen and risk of clinical malaria: Implications for vaccine development. Sci. Transl. Med. 2009, 1, 2ra5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neafsey, D.E.; Schaffner, S.F.; Volkman, S.K.; Park, D.; Montgomery, P.; Milner, D.A., Jr.; Lukens, A.; Rosen, D.; Daniels, R.; Houde, N.; et al. Genome-wide SNP genotyping highlights the role of natural selection in Plasmodium falciparum population divergence. Genome Biol. 2008, 9, R171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, K.A.; Drabek, E.F.; Dwivedi, A.; Stucke, E.M.; Crabtree, J.; Dara, A.; Shah, Z.; Adams, M.; Li, T.; Rodrigues, P.T.; et al. Strains used in whole organism Plasmodium falciparum vaccine trials differ in genome structure, sequence, and immunogenic potential. Genome Med. 2020, 12, 6. [Google Scholar] [CrossRef] [Green Version]
- Adepoju, P. RTS,S malaria vaccine pilots in three African countries. Lancet 2019, 393, 1685. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 1–33. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J. DNA sequence polymorphism analysis using DnaSP. Methods Mol. Biol. 2009, 537, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Lyke, K.E.; Fernandez-Vina, M.A.; Cao, K.; Hollenbach, J.; Coulibaly, D.; Kone, A.K.; Guindo, A.; Burdett, L.A.; Hartzman, R.J.; Wahl, A.R.; et al. Association of HLA alleles with Plasmodium falciparum severity in Malian children. Tissue Antigens 2011, 77, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Moormann, A.M.; Lyke, K.E.; Masaberg, C.; Sumba, O.P.; Doumbo, O.K.; Koech, D.; Lancaster, A.; Nelson, M.; Meyer, D.; et al. Differentiation between African populations is evidenced by the diversity of alleles and haplotypes of HLA class I loci. Tissue Antigens 2004, 63, 293–325. [Google Scholar] [CrossRef]
- Sette, A.; Sidney, J. Nine major HLA class I supertypes account for the vast preponderance of HLA-A and -B polymorphism. Immunogenetics 1999, 50, 201–212. [Google Scholar] [CrossRef]
- Sette, A.; Sidney, J. HLA supertypes and supermotifs: A functional perspective on HLA polymorphism. Curr. Opin. Immunol. 1998, 10, 478–482. [Google Scholar] [CrossRef]
- Sette, A.; Vitiello, A.; Reherman, B.; Fowler, P.; Nayersina, R.; Kast, W.M.; Melief, C.J.; Oseroff, C.; Yuan, L.; Ruppert, J.; et al. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J. Immunol. 1994, 153, 5586–5592. [Google Scholar]
- Rubinstein, N.D.; Mayrose, I.; Halperin, D.; Yekutieli, D.; Gershoni, J.M.; Pupko, T. Computational characterization of B-cell epitopes. Mol. Immunol. 2008, 45, 3477–3489. [Google Scholar] [CrossRef] [Green Version]
- Sivalingam, G.N.; Shepherd, A.J. An analysis of B-cell epitope discontinuity. Mol. Immunol. 2012, 51, 304–309. [Google Scholar] [CrossRef]
- Ansari, H.R.; Raghava, G.P. Identification of conformational B-cell Epitopes in an antigen from its primary sequence. Immunome Res. 2010, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Roy, A.; Zhang, Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Peng, Z.; Zhang, Y.; Yang, J. COACH-D: Improved protein-ligand binding sites prediction with refined ligand-binding poses through molecular docking. Nucleic Acids Res. 2018, 46, W438–W442. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Yang, J.; Zhang, Y. COFACTOR: An accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 2012, 40, W471–W477. [Google Scholar] [CrossRef] [Green Version]
- Brylinski, M.; Skolnick, J. A threading-based method (FINDSITE) for ligand-binding site prediction and functional annotation. Proc. Natl. Acad. Sci. USA 2008, 105, 129–134. [Google Scholar] [CrossRef] [Green Version]
- Capra, J.A.; Laskowski, R.A.; Thornton, J.M.; Singh, M.; Funkhouser, T.A. Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure. PLoS Comput. Biol. 2009, 5, e1000585. [Google Scholar] [CrossRef] [Green Version]
- Ouattara, A.; Barry, A.E.; Dutta, S.; Remarque, E.J.; Beeson, J.G.; Plowe, C.V. Designing malaria vaccines to circumvent antigen variability. Vaccine 2015, 33, 7506–7512. [Google Scholar] [CrossRef] [Green Version]
- Ouattara, A.; Mu, J.; Takala-Harrison, S.; Saye, R.; Sagara, I.; Dicko, A.; Niangaly, A.; Duan, J.; Ellis, R.D.; Miller, L.H.; et al. Lack of allele-specific efficacy of a bivalent AMA1 malaria vaccine. Malar. J. 2010, 9, 175. [Google Scholar] [CrossRef] [Green Version]
- Ouattara, A.; Niangaly, A.; Adams, M.; Coulibaly, D.; Kone, A.K.; Traore, K.; Laurens, M.B.; Tolo, Y.; Kouriba, B.; Diallo, D.A.; et al. Epitope-based sieve analysis of Plasmodium falciparum sequences from a FMP2.1/AS02A vaccine trial is consistent with differential vaccine efficacy against immunologically relevant AMA1 variants. Vaccine 2020, 38, 5700–5706. [Google Scholar] [CrossRef]
- Polley, S.D.; Tetteh, K.K.; Lloyd, J.M.; Akpogheneta, O.J.; Greenwood, B.M.; Bojang, K.A.; Conway, D.J. Plasmodium falciparum merozoite surface protein 3 is a target of allele-specific immunity and alleles are maintained by natural selection. J. Infect. Dis. 2007, 195, 279–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.M.; Moon, S.U.; Kim, J.Y.; Cho, S.H.; Lin, K.; Sohn, W.M.; Kim, T.S.; Na, B.K. Genetic polymorphism of merozoite surface protein-1 and merozoite surface protein-2 in Plasmodium falciparum field isolates from Myanmar. Malar. J. 2010, 9, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, S.S.; Diekema, D.J.; Heilmann, K.P.; Dohrn, C.L.; Riahi, F.; Doern, G.V. Changes in pneumococcal serotypes and antimicrobial resistance after introduction of the 13-valent conjugate vaccine in the United States. Antimicrob. Agents Chemother. 2014, 58, 6484–6489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyke, K.E.; Diallo, D.A.; Dicko, A.; Kone, A.; Coulibaly, D.; Guindo, A.; Cissoko, Y.; Sangare, L.; Coulibaly, S.; Dakouo, B.; et al. Association of intraleukocytic Plasmodium falciparum malaria pigment with disease severity, clinical manifestations, and prognosis in severe malaria. Am. J. Trop. Med. Hyg. 2003, 69, 253–259. [Google Scholar] [CrossRef]
- A global network for investigating the genomic epidemiology of malaria. Nature 2008, 456, 732–737. [CrossRef]
- Duffy, P.E.; Sahu, T.; Akue, A.; Milman, N.; Anderson, C. Pre-erythrocytic malaria vaccines: Identifying the targets. Expert Rev. Vaccines 2012, 11, 1261–1280. [Google Scholar] [CrossRef] [Green Version]
- Taylor-Robinson, A.W. Immunity to liver stage malaria: Considerations for vaccine design. Immunol. Res. 2003, 27, 53–70. [Google Scholar] [CrossRef]
- Sauerwein, R.W. The first malaria vaccine: A significant milestone. Ned. Tijdschr. Geneeskd. 2015, 159, A9730. [Google Scholar]
- Mahmoudi, S.; Keshavarz, H. Efficacy of Phase 3 Trial of RTS, S/AS01 Malaria Vaccine in infants: A systematic review and meta-analysis. Hum. Vaccines Immunother. 2017, 11, 261. [Google Scholar] [CrossRef]
- Rosenthal, P.J. The RTS,S/AS01 vaccine continues to show modest protection against malaria in African infants and children. Evid. Based Med. 2015, 20, 179. [Google Scholar] [CrossRef]
- Rampling, T.; Ewer, K.J.; Bowyer, G.; Edwards, N.J.; Wright, D.; Sridhar, S.; Payne, R.; Powlson, J.; Bliss, C.; Venkatraman, N.; et al. Safety and efficacy of novel malaria vaccine regimens of RTS,S/AS01B alone, or with concomitant ChAd63-MVA-vectored vaccines expressing ME-TRAP. NPJ Vaccines 2018, 3, 49. [Google Scholar] [CrossRef] [PubMed]
- Bowyer, G.; Grobbelaar, A.; Rampling, T.; Venkatraman, N.; Morelle, D.; Ballou, R.W.; Hill, A.V.S.; Ewer, K.J. CXCR3(+) T Follicular Helper Cells Induced by Co-Administration of RTS,S/AS01B and Viral-Vectored Vaccines Are Associated With Reduced Immunogenicity and Efficacy Against Malaria. Front. Immunol. 2018, 9, 660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, K.A.; Snaith, R.; Cottingham, M.G.; Gilbert, S.C.; Hill, A.V.S. Enhancing protective immunity to malaria with a highly immunogenic virus-like particle vaccine. Sci. Rep. 2017, 7, 46621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halbroth, B.R.; Sebastian, S.; Salman, A.M.; Ulaszewska, M.; Gola, A.; Longley, R.J.; Janse, C.J.; Khan, S.M.; Hill, A.V.S.; Spencer, A.J. Preclinical Development and Assessment of Viral Vectors Expressing a Fusion Antigen of Plasmodium falciparum LSA1 and LSAP2 for Efficacy against Liver-Stage Malaria. Infect. Immun. 2020, 88, e00573-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longley, R.J.; Halbroth, B.R.; Salman, A.M.; Ewer, K.J.; Hodgson, S.H.; Janse, C.J.; Khan, S.M.; Hill, A.V.S.; Spencer, A.J. Assessment of the Plasmodium falciparum Preerythrocytic Antigen UIS3 as a Potential Candidate for a Malaria Vaccine. Infect. Immun. 2017, 85, e00641-16. [Google Scholar] [CrossRef] [Green Version]
- Kimani, D.; Jagne, Y.J.; Cox, M.; Kimani, E.; Bliss, C.M.; Gitau, E.; Ogwang, C.; Afolabi, M.O.; Bowyer, G.; Collins, K.A.; et al. Translating the immunogenicity of prime-boost immunization with ChAd63 and MVA ME-TRAP from malaria naive to malaria-endemic populations. Mol. Ther. 2014, 22, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Beeson, J.G.; Kurtovic, L.; Dobano, C.; Opi, D.H.; Chan, J.A.; Feng, G.; Good, M.F.; Reiling, L.; Boyle, M.J. Challenges and strategies for developing efficacious and long-lasting malaria vaccines. Sci. Transl. Med. 2019, 11, eaau1458. [Google Scholar] [CrossRef]
- Patarroyo, M.E.; Arevalo-Pinzon, G.; Reyes, C.; Moreno-Vranich, A.; Patarroyo, M.A. Malaria Parasite Survival Depends on Conserved Binding Peptides’ Critical Biological Functions. Curr. Issues Mol. Biol. 2016, 18, 57–78. [Google Scholar]
- Mackellar, D.C.; O’Neill, M.T.; Aly, A.S.; Sacci, J.B.; Cowman, A.F., Jr.; Kappe, S.H. Plasmodium falciparum PF10_0164 (ETRAMP10.3) is an essential parasitophorous vacuole and exported protein in blood stages. Eukaryot. Cell 2010, 9, 784–794. [Google Scholar] [CrossRef] [Green Version]
- Ajibola, O.; Diop, M.F.; Ghansah, A.; Amenga-Etego, L.; Golassa, L.; Apinjoh, T.; Randrianarivelojosia, M.; Maiga-Ascofare, O.; Yavo, W.; Bouyou-Akotet, M.; et al. In silico characterisation of putative Plasmodium falciparum vaccine candidates in African malaria populations. Sci. Rep. 2021, 11, 16215. [Google Scholar] [CrossRef]
- Manske, M.; Miotto, O.; Campino, S.; Auburn, S.; Almagro-Garcia, J.; Maslen, G.; O’Brien, J.; Djimde, A.; Doumbo, O.; Zongo, I.; et al. Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature 2012, 487, 375–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. World Malaria Report 2021; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Hughes, A.L.; Verra, F. Very large long-term effective population size in the virulent human malaria parasite Plasmodium falciparum. Proc. Biol. Sci. 2001, 268, 1855–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sondo, P.; Derra, K.; Lefevre, T.; Diallo-Nakanabo, S.; Tarnagda, Z.; Zampa, O.; Kazienga, A.; Valea, I.; Sorgho, H.; Ouedraogo, J.B.; et al. Genetically diverse Plasmodium falciparum infections, within-host competition and symptomatic malaria in humans. Sci. Rep. 2019, 9, 127. [Google Scholar] [CrossRef] [PubMed]
- Abukari, Z.; Okonu, R.; Nyarko, S.B.; Lo, A.C.; Dieng, C.C.; Salifu, S.P.; Gyan, B.A.; Lo, E.; Amoah, L.E. The Diversity, Multiplicity of Infection and Population Structure of P. falciparum Parasites Circulating in Asymptomatic Carriers Living in High and Low Malaria Transmission Settings of Ghana. Genes 2019, 10, 434. [Google Scholar] [CrossRef] [Green Version]
- Nascutiu, A.M. “Hide-and-seek” as modus vivendi of malaria parasites. Rom. Arch. Microbiol. Immunol. 2002, 61, 301–314. [Google Scholar]
- Park, A.W.; Daly, J.M.; Lewis, N.S.; Smith, D.J.; Wood, J.L.; Grenfell, B.T. Quantifying the impact of immune escape on transmission dynamics of influenza. Science 2009, 326, 726–728. [Google Scholar] [CrossRef] [Green Version]
- Campeotto, I.; Goldenzweig, A.; Davey, J.; Barfod, L.; Marshall, J.M.; Silk, S.E.; Wright, K.E.; Draper, S.J.; Higgins, M.K.; Fleishman, S.J. One-step design of a stable variant of the malaria invasion protein RH5 for use as a vaccine immunogen. Proc. Natl. Acad. Sci. USA 2017, 114, 998–1002. [Google Scholar] [CrossRef] [Green Version]
- Patarroyo, M.E.; Cifuentes, G.; Bermudez, A.; Patarroyo, M.A. Strategies for developing multi-epitope, subunit-based, chemically synthesized anti-malarial vaccines. J. Cell. Mol. Med. 2008, 12, 1915–1935. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.M.; Wu, C.K.; Plieskatt, J.; McAdams, D.H.; Miura, K.; Ockenhouse, C.; King, C.R. Assessment of Pfs25 expressed from multiple soluble expression platforms for use as transmission-blocking vaccine candidates. Malar. J. 2016, 15, 405. [Google Scholar] [CrossRef] [Green Version]
- Thompson, C.M.; Petiot, E.; Mullick, A.; Aucoin, M.G.; Henry, O.; Kamen, A.A. Critical assessment of influenza VLP production in Sf9 and HEK293 expression systems. BMC Biotechnol. 2015, 15, 31. [Google Scholar] [CrossRef] [Green Version]
- Doolan, D.L.; Hoffman, S.L. The complexity of protective immunity against liver-stage malaria. J. Immunol. 2000, 165, 1453–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, L.H.; Sano, G.; Hafalla, J.C.; Morrot, A.; Curotto de Lafaille, M.A.; Zavala, F. IL-4-secreting CD4+ T cells are crucial to the development of CD8+ T-cell responses against malaria liver stages. Nat. Med. 2002, 8, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Overstreet, M.G.; Chen, Y.C.; Cockburn, I.A.; Tse, S.W.; Zavala, F. CD4+ T cells modulate expansion and survival but not functional properties of effector and memory CD8+ T cells induced by malaria sporozoites. PLoS ONE 2011, 6, e15948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schofield, L.; Villaquiran, J.; Ferreira, A.; Schellekens, H.; Nussenzweig, R.; Nussenzweig, V. Gamma interferon, CD8+ T cells and antibodies required for immunity to malaria sporozoites. Nature 1987, 330, 664–666. [Google Scholar] [CrossRef]
- Helg, A.; Mueller, M.S.; Joss, A.; Poltl-Frank, F.; Stuart, F.; Robinson, J.A.; Pluschke, G. Comparison of analytical methods for the evaluation of antibody responses against epitopes of polymorphic protein antigens. J. Immunol. Methods 2003, 276, 19–31. [Google Scholar] [CrossRef]
- Terasaki, P.I. A brief history of HLA. Immunol. Res. 2007, 38, 139–148. [Google Scholar] [CrossRef]
- Zhao, W.; Sher, X. Systematically benchmarking peptide-MHC binding predictors: From synthetic to naturally processed epitopes. PLoS Comput. Biol. 2018, 14, e1006457. [Google Scholar] [CrossRef]
- Sanchez-Trincado, J.L.; Gomez-Perosanz, M.; Reche, P.A. Fundamentals and Methods for T- and B-Cell Epitope Prediction. J. Immunol. Res. 2017, 2017, 2680160. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigens | Hd | π Per 100 bp | Breaks in Sequence Conservation across Haplotypes |
|---|---|---|---|
| SAP1 | 0.9990 | 0.181 | 58/2940 |
| LSA3 | 0.9989 | 0.358 | 16/1558 |
| CSP | 0.9952 | 0.940 | 2/397 |
| CelTOS | 0.9884 | 1.530 | 3/182 |
| SPECT2 | 0.9856 | 0.194 | 7/842 |
| LSA1 | 0.9845 | 0.338 | 10/1162 |
| LSA3 | 0.9989 | 0.358 | 5/478 |
| UIS4 | 0.7190 | 0.451 | 1/108 |
| P36 | 0.4100 | 0.071 | 2/379 |
| UIS3 | 0.2100 | 0.038 | 2/229 |
| Antigen | Strong-Binding CD4+ Epitope | Relative Surface Accessibility CD4+ | Strong-Binding CD8+ Epitope | Relative Surface Accessibility CD8+ |
|---|---|---|---|---|
| CELTOS | MNALRRLPVICS | Exposed | LPVICSFLVF | Exposed |
| CSP | KLAILSVSSFLF | Exposed | - | - |
| LAILSVSSFLFV | Exposed | SSFLFVEALF * | Exposed | |
| ENWYSLKKNSRS | Exposed | - | - | |
| LSA1 | TNFKSLLRNLGV | Buried | - | - |
| NFKSLLRNLGVS | Buried | - | - | |
| QTNFKSLLRNLG | Buried | - | - | |
| FKSLLRNLGVSE | Buried | KFIKSLFHIF * | Buried | |
| NFKSLLRNLGVS | Buried | - | - | |
| TNFKSLLRNLGV | Buried | - | - | |
| ISFYFILVNLLI | Buried | - | - | |
| SFYFILVNLLIF | Buried | - | - | |
| LSA3 | None | - | ASYVVGFFTF * | Buried |
| - | - | - | SYVVGFFTFS * | Buried |
| - | - | - | PFYSFVFDIF * | Buried |
| - | - | - | KVKNFVKKYK | Exposed |
| LSA3 | - | - | KVDKNNKVPK * | Exposed |
| - | - | - | KTRKKAQRPK * | Buried |
| - | - | - | KVFAAPFISA * | Buried |
| - | - | - | KINKYFFLIK | Exposed |
| - | IRYNKSRSSNLI | Buried | - | - |
| - | AIRYNKSRSSNL | Buried | - | - |
| - | KFVQLLKSIDIK | Buried | - | - |
| - | RYNKSRSSNLIA | Buried | - | - |
| P36 | FVQLLKSIDIKM | Buried | - | - |
| - | AIRYNKSRSSNL | Buried | KSKCETKIEK | Buried |
| - | EAIRYNKSRSSN | Buried | - | - |
| - | EEAIRYNKSRSS | Buried | - | - |
| - | IRYNKSRSSNLI | Buried | - | - |
| - | SKFVQLLKSIDI | Buried | - | - |
| - | MCYHFTMKRKKL | Exposed | - | - |
| - | HMCYHFTMKRKK | Exposed | - | - |
| - | NLFGLSSSKYIL | Buried | - | - |
| - | QNLFGLSSSKYI | Exposed | - | - |
| - | NININFVCSNVI | Buried | KYILFNNFLI | Buried |
| - | ININFVCSNVIQ | Buried | ILFNNFLILF * | Buried |
| P52 | CYHFTMKRKKLF | Exposed | VYFIFLSFII * | Exposed |
| - | YHFTMKRKKLFV | Exposed | KVKHIMRINI | Buried |
| - | LFGLSSSKYILF | Buried | RTRTFWQNLF | Exposed |
| - | GTMIIYTKNINS | Buried | KLSRNHSFSS | Buried |
| - | MIIYTKNINSLM | Buried | NPSNCFHDVY | Buried |
| - | TMIIYTKNINSL | Buried | - | - |
| - | VGTMIIYTKNIN | Buried | - | - |
| - | FGLSSSKYILFN | Buried | - | - |
| - | - | - | VKYFNKPIQF | Exposed |
| - | - | - | YKYIQNIILF | Buried |
| - | - | - | YFMPKNDLNF | Buried |
| - | - | - | KYIQNIILFL | Buried |
| - | - | - | NYMPQNYYHI | Buried |
| SAP1 | None | - | RIFFSFFSYF | Buried |
| - | - | - | RFKLTCNFKF | Buried |
| - | - | - | KLKNFFLNYK | Buried |
| - | - | - | KMTKNYNINA | Exposed |
| - | - | - | YTRAVWLLKK | Buried |
| - | - | - | MPKNDLNFIF | Buried |
| - | - | - | MPQNYYHINY | Buried |
| - | KLRILKKHYYVV * | Exposed | LYFIGIGYNL | Buried |
| - | LRILKKHYYVVF * | Exposed | IYVLCVDTTI | Buried |
| SPECT2 | MKLRILKKHYYV * | Exposed | KRSKKTFLVK | Buried |
| - | MKLRILKKHYYV * | Exposed | KVVMFGFSLK | Buried |
| - | KLRILKKHYYVV * | Exposed | RSKKTFLVKS | Buried |
| - | LRILKKHYYVVF * | Exposed | KKIKHSFNLA | Exposed |
| - | - | - | YIPWDKTTAY | Buried |
| - | - | - | - | - |
| - | - | - | - | - |
| - | KYHLQKVYANYL * | Buried | - | - |
| - | YHLQKVYANYLS * | Buried | - | - |
| - | MEYMQKFVQGLQ * | Buried | - | - |
| - | NMEYMQKFVQGL * | Buried | - | - |
| UIS3 | NNMEYMQKFVQG * | Buried | None | - |
| - | VNNMEYMQKFVQ * | Buried | - | - |
| - | LIYYSLIASGAI * | Exposed | - | - |
| - | IYYSLIASGAIA * | Exposed | - | - |
| KQIIISTAAVAL * | Exposed | - | - | |
| QIIISTAAVALA * | Exposed | - | - | |
| UIS4 | RTLEKLLRKKQI * | Exposed | None | - |
| DRTLEKLLRKKQ | Exposed | - | - | |
| LEKLLRKKQII * | Exposed | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouattara, A.; Dwivedi, A.; Adams, M.; Niangaly, A.; Laurens, M.B.; Nyunt, M.M.; Plowe, C.V.; Djimde, A.; Takala-Harrison, S.; Silva, J.C. An In Silico Analysis of Malaria Pre-Erythrocytic-Stage Antigens Interpreting Worldwide Genetic Data to Suggest Vaccine Candidate Variants and Epitopes. Microorganisms 2022, 10, 1090. https://doi.org/10.3390/microorganisms10061090
Ouattara A, Dwivedi A, Adams M, Niangaly A, Laurens MB, Nyunt MM, Plowe CV, Djimde A, Takala-Harrison S, Silva JC. An In Silico Analysis of Malaria Pre-Erythrocytic-Stage Antigens Interpreting Worldwide Genetic Data to Suggest Vaccine Candidate Variants and Epitopes. Microorganisms. 2022; 10(6):1090. https://doi.org/10.3390/microorganisms10061090
Chicago/Turabian StyleOuattara, Amed, Ankit Dwivedi, Matthew Adams, Amadou Niangaly, Matthew B. Laurens, Myaing M. Nyunt, Christopher V. Plowe, Abdoulaye Djimde, Shannon Takala-Harrison, and Joana C. Silva. 2022. "An In Silico Analysis of Malaria Pre-Erythrocytic-Stage Antigens Interpreting Worldwide Genetic Data to Suggest Vaccine Candidate Variants and Epitopes" Microorganisms 10, no. 6: 1090. https://doi.org/10.3390/microorganisms10061090
APA StyleOuattara, A., Dwivedi, A., Adams, M., Niangaly, A., Laurens, M. B., Nyunt, M. M., Plowe, C. V., Djimde, A., Takala-Harrison, S., & Silva, J. C. (2022). An In Silico Analysis of Malaria Pre-Erythrocytic-Stage Antigens Interpreting Worldwide Genetic Data to Suggest Vaccine Candidate Variants and Epitopes. Microorganisms, 10(6), 1090. https://doi.org/10.3390/microorganisms10061090