
Effect of Caging on Cryptosporidium parvum Proliferation in Mice


Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Experiments
2.2. Quantification of Fecal Oocysts
2.3. 16S Amplicon Sequencing, Quality Control and Bioinformatics
3. Results
3.1. Effect of Caging
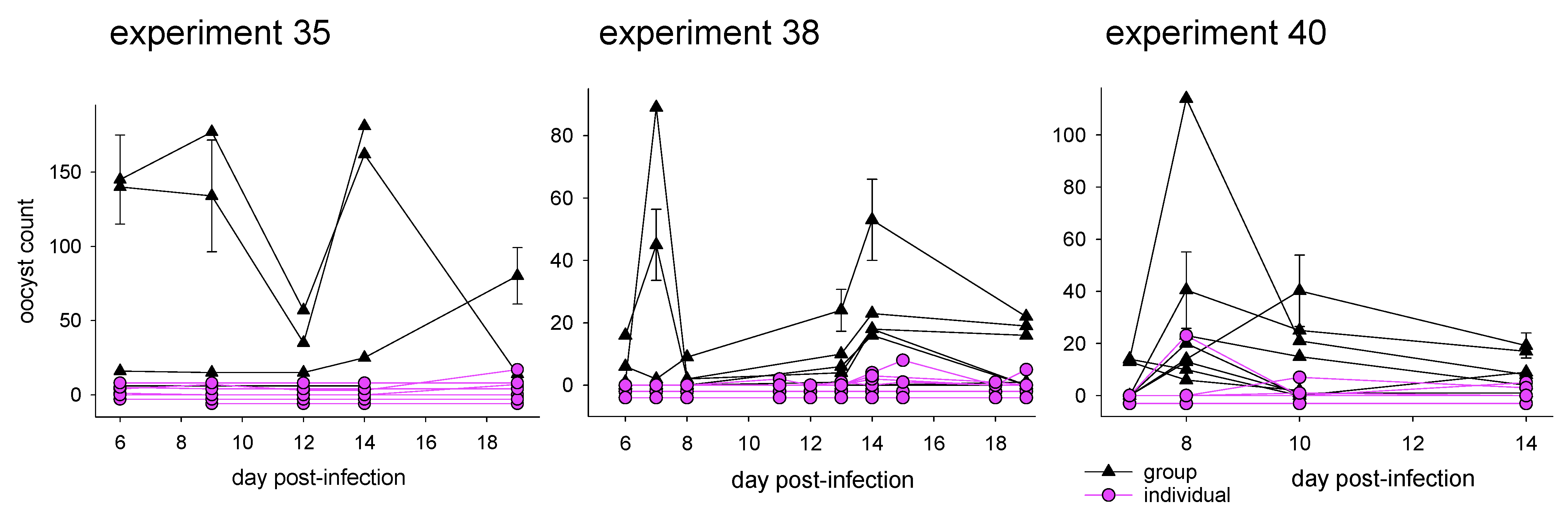
3.1.1. Effect of Caging on Cryptosporidiosis
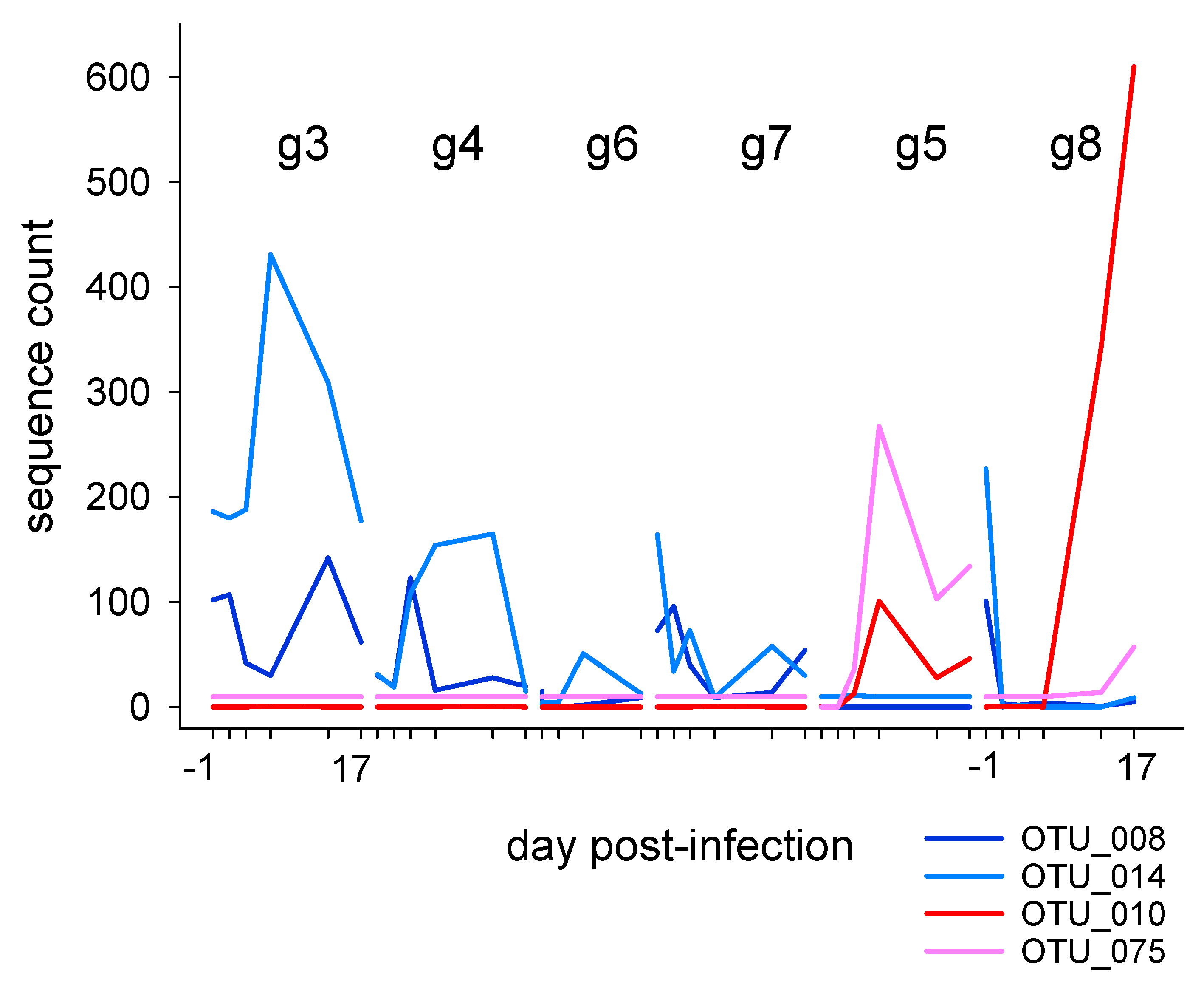
3.1.2. Effect of Caging on Fecal Microbiota
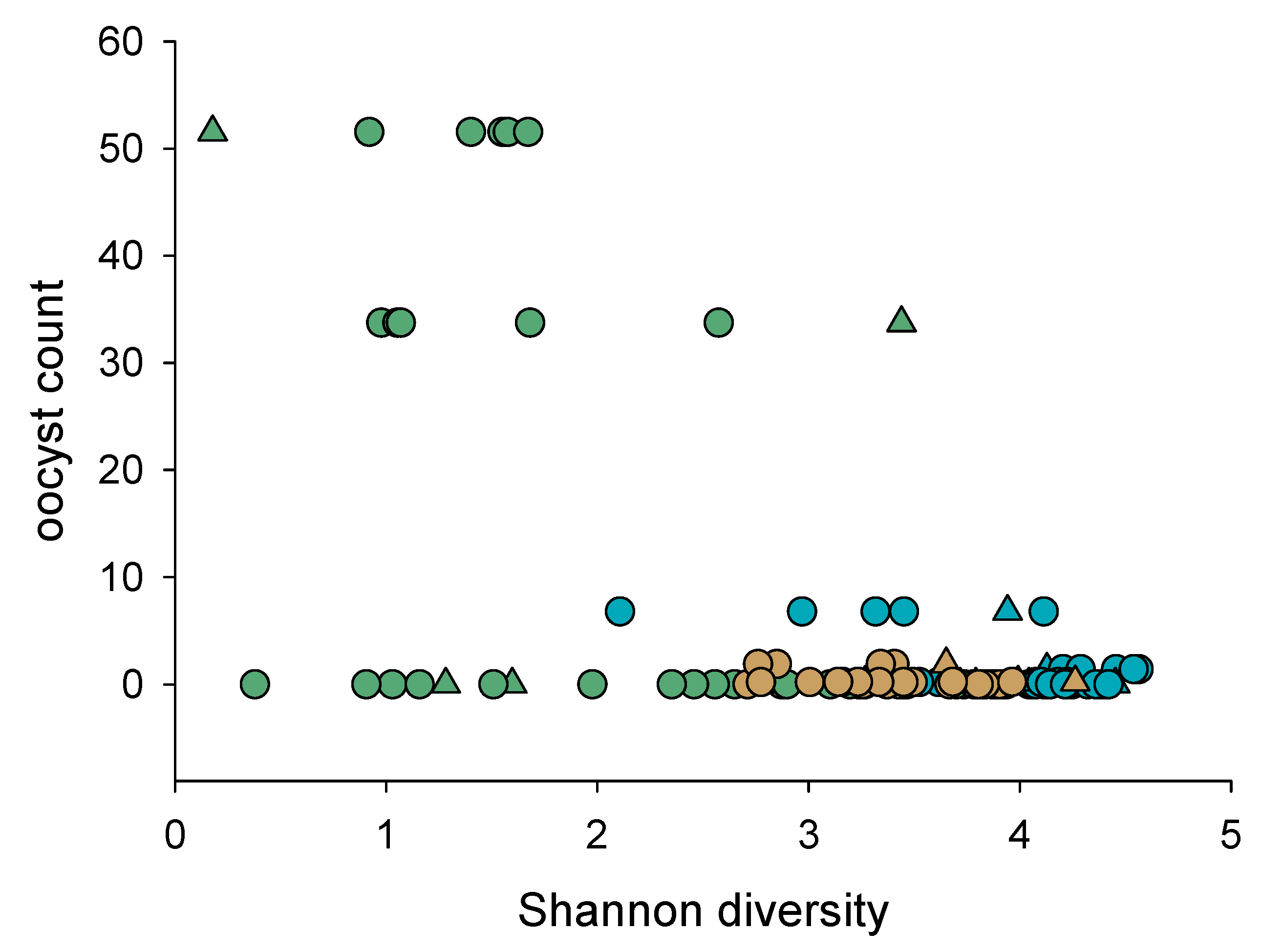
3.1.3. Association of Cryptosporidiosis and Fecal Microbiota
3.2. Mice caged Individually
3.2.1. Fecal Microbiota in Individually Caged Mice
3.2.2. Association of Cryptosporidiosis and Fecal Microbiota in Individually Caged Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kotloff, K.L.; Blackwelder, W.C.; Nasrin, D.; Nataro, J.P.; Farag, T.H.; van Eijk, A.; Adegbola, R.A.; Alonso, P.L.; Breiman, R.F.; Faruque, A.S.; et al. The Global Enteric Multicenter Study (GEMS) of diarrheal disease in infants and young children in developing countries: Epidemiologic and clinical methods of the case/control study. Clin. Infect. Dis. 2012, 55 (Suppl. 4), S232–S245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Checkley, W.; White, A.C., Jr.; Jaganath, D.; Arrowood, M.J.; Chalmers, R.M.; Chen, X.M.; Fayer, R.; Griffiths, J.K.; Guerrant, R.L.; Hedstrom, L.; et al. A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for Cryptosporidium. Lancet Infect. Dis. 2015, 15, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Striepen, B. Parasitic infections: Time to tackle cryptosporidiosis. Nature 2013, 503, 189–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shikani, H.; Weiss, L.M. Human cryptosporidiosis: A clinical perspective. In Cryptosporidium: Parasite and Disease; Springer: Berlin/Heidelberg, Germany, 2014; pp. 383–421. [Google Scholar]
- Morada, M.; Lee, S.; Gunther-Cummins, L.; Weiss, L.M.; Widmer, G.; Tzipori, S.; Yarlett, N. Continuous culture of Cryptosporidium parvum using hollow fiber technology. Int. J. Parasitol. 2015, 46, 21–29. [Google Scholar] [CrossRef]
- Wilke, G.; Funkhouser-Jones, L.J.; Wang, Y.; Ravindran, S.; Wang, Q.; Beatty, W.L.; Baldridge, M.T.; Van Dussen, K.L.; Shen, B.; Kuhlenschmidt, M.S.; et al. A Stem-Cell-Derived Platform Enables Complete Cryptosporidium Development In Vitro and Genetic Tractability. Cell Host Microbe 2019, 26, 123–134.e8. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, B.C.M.; Widmer, G. Probiotic product enhances susceptibility of mice to cryptosporidiosis. Appl. Environ. Microbiol. 2018, 84, e01408-18. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, B.C.M.; Bresciani, K.D.S.; Widmer, G. Deprivation of dietary fiber enhances susceptibility of mice to cryptosporidiosis. PLoS Negl. Trop. Dis. 2019, 13, e0007411. [Google Scholar] [CrossRef] [Green Version]
- Berry, A.S.; Johnson, K.; Martins, R.; Sullivan, M.C.; Farias Amorim, C.; Putre, A.; Scott, A.; Wang, S.; Lindsay, B.; Baldassano, R.N. Natural infection with Giardia is associated with altered community structure of the human and canine gut microbiome. Msphere 2020, 5, e00670-20. [Google Scholar] [CrossRef]
- Watanabe, K.; Petri, W.A., Jr. Molecular biology research to benefit patients with Entamoeba histolytica infection. Mol. Microbiol. 2015, 98, 208–217. [Google Scholar] [CrossRef]
- Madlala, T.; Okpeku, M.; Adeleke, M.A. Understanding the interactions between Eimeria infection and gut microbiota, towards the control of chicken coccidiosis: A review. Parasite 2021, 28, 48. [Google Scholar] [CrossRef]
- Shen, H.; Fan, X.; Qiao, Y.; Jiang, G.; Wan, X.; Cheng, J.; Li, H.; Dou, Y.; Li, H.; Wang, L.; et al. The links among Enterocytozoon hepatopenaei infection, growth retardation and intestinal microbiota in different sized shrimp Penaeus vannamei. Aquac. Rep. 2021, 21, 100888. [Google Scholar] [CrossRef]
- Leung, J.M.; Graham, A.L.; Knowles, S.C.L. Parasite-Microbiota Interactions with the Vertebrate Gut: Synthesis Through an Ecological Lens. Front. Microbiol. 2018, 9, 843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Striepen, B.; Kissinger, J.C. Genomics meets transgenics in search of the elusive Cryptosporidium drug target. Trends Parasitol. 2004, 20, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Striepen, B.; Pruijssers, A.J.; Huang, J.; Li, C.; Gubbels, M.J.; Umejiego, N.N.; Hedstrom, L.; Kissinger, J.C. Gene transfer in the evolution of parasite nucleotide biosynthesis. Proc. Natl. Acad. Sci. USA 2004, 101, 3154–3159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuckin, M.A.; Linden, S.K.; Sutton, P.; Florin, T.H. Mucin dynamics and enteric pathogens. Nat. Rev. Microbiol. 2011, 9, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Backhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harp, J.A.; Chen, W.; Harmsen, A.G. Resistance of severe combined immunodeficient mice to infection with Cryptosporidium parvum: The importance of intestinal microflora. Infect. Immun. 1992, 60, 3509–3512. [Google Scholar] [CrossRef] [Green Version]
- Harp, J.A. Cryptosporidium and host resistance: Historical perspective and some novel approaches. Anim. Health Res. Rev. 2003, 4, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Lacroix-Lamande, S.; Guesdon, W.; Drouet, F.; Potiron, L.; Lantier, L.; Laurent, F. The gut flora is required for the control of intestinal infection by poly(I:C) administration in neonates. Gut Microbes 2014, 5, 533–540. [Google Scholar] [CrossRef]
- Lantier, L.; Drouet, F.; Guesdon, W.; Mancassola, R.; Metton, C.; Lo-Man, R.; Werts, C.; Laurent, F.; Lacroix-Lamande, S. Poly(I:C)-induced protection of neonatal mice against intestinal Cryptosporidium parvum infection requires an additional TLR5 signal provided by the gut flora. J. Infect. Dis. 2014, 209, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Gorla, S.K.; McNair, N.N.; Yang, G.; Gao, S.; Hu, M.; Jala, V.R.; Haribabu, B.; Striepen, B.; Cuny, G.D.; Mead, J.R.; et al. Validation of IMP dehydrogenase inhibitors in a mouse model of cryptosporidiosis. Antimicrob. Agents Chemother. 2014, 58, 1603–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanad, M.M.; Al-Malki, J.S.; Al-Ghabban, A.G. Control of cryptosporidiosis by probiotic bacteria. In Proceedings of the International Conference on Agricultural, Ecological and Medical Sciences (AEMS-2015), Phuket, Thailand, 7–8 April 2015; pp. 7–8. [Google Scholar]
- Alak, J.I.; Wolf, B.W.; Mdurvwa, E.G.; Pimentel-Smith, G.E.; Adeyemo, O. Effect of Lactobacillus reuteri on intestinal resistance to Cryptosporidium parvum infection in a murine model of acquired immunodeficiency syndrome. J. Infect. Dis. 1997, 175, 218–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alak, J.; Wolf, B.; Mdurvwa, E.; Pimentel-Smith, G.; Kolavala, S.; Abdelrahman, H.; Suppiramaniam, V. Supplementation with Lactobacillus reuteri or L. acidophilus reduced intestinal shedding of Cryptosporidium parvum oocysts in immunodeficient C57BL/6 mice. Cell. Mol. Biol. 1999, 45, 855–863. [Google Scholar]
- Waters, W.; Harp, J.; Wannemuehler, M.; Carbajal, N.; Casas, I. Effects of Lactobacillus reuteri on Cryptosporidium parvum infection of gnotobiotic TCR-alpha-deficient mice. J. Eukaryot. Microbiol. 1999, 46, 60S–61S. [Google Scholar] [PubMed]
- Foster, J.C.; Glass, M.D.; Courtney, P.D.; Ward, L.A. Effect of Lactobacillus and Bifidobacterium on Cryptosporidium parvum oocyst viability. Food Microbiol. 2003, 20, 351–357. [Google Scholar] [CrossRef]
- Del Coco, V.F.; Sparo, M.D.; Sidoti, A.; Santin, M.; Basualdo, J.A.; Cordoba, M.A. Effects of Enterococcus faecalis CECT 7121 on Cryptosporidium parvum infection in mice. Parasitol. Res. 2016, 115, 3239–3244. [Google Scholar] [CrossRef]
- Charania, R.; Wade, B.E.; McNair, N.N.; Mead, J.R. Changes in the Microbiome of Cryptosporidium-Infected Mice Correlate to Differences in Susceptibility and Infection Levels. Microorganisms 2020, 8, 879. [Google Scholar] [CrossRef]
- Mammeri, M.; Chevillot, A.; Thomas, M.; Julien, C.; Auclair, E.; Pollet, T.; Polack, B.; Vallée, I.; Adjou, K.T. Cryptosporidium parvum-Infected Neonatal Mice Show Gut Microbiota Remodelling Using High-Throughput Sequencing Analysis: Preliminary Results. Acta Parasitol. 2019, 64, 268–275. [Google Scholar] [CrossRef]
- Ras, R.; Huynh, K.; Desoky, E.; Badawy, A.; Widmer, G. Perturbation of the intestinal microbiota of mice infected with Cryptosporidium parvum. Int. J. Parasitol. 2015, 45, 567–573. [Google Scholar] [CrossRef]
- Weber, E.M.; Dallaire, J.A.; Gaskill, B.N.; Pritchett-Corning, K.R.; Garner, J.P. Aggression in group-housed laboratory mice: Why can’t we solve the problem? Lab Anim. 2017, 46, 157–161. [Google Scholar] [CrossRef] [Green Version]
- Langgartner, D.; Füchsl, A.M.; Uschold-Schmidt, N.; Slattery, D.A.; Reber, S.O. Chronic subordinate colony housing paradigm: A mouse model to characterize the consequences of insufficient glucocorticoid signaling. Front. Psychiatry 2015, 6, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freestone, P.P.E.; Sandrini, S.M.; Haigh, R.D.; Lyte, M. Microbial endocrinology: How stress influences susceptibility to infection. Trends Microbiol. 2008, 16, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Healey, M.C. The immunosuppressive effects of dexamethasone administered in drinking water to C57BL/6N mice infected with Cryptosporidium parvum. J. Parasitol. 1993, 79, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Widmer, G.; Feng, X.; Tanriverdi, S. Genotyping of Cryptosporidium parvum with microsatellite markers. Methods Mol. Biol. 2004, 268, 177–187. [Google Scholar] [PubMed]
- Ma, P.; Soave, R. Three-step stool examination for cryptosporidiosis in 10 homosexual men with protracted watery diarrhea. J. Infect. Dis. 1983, 147, 824–828. [Google Scholar] [CrossRef]
- Fontaine, M.; Guillot, E. Development of a TaqMan quantitative PCR assay specific for Cryptosporidium parvum. FEMS Microbiol. Lett. 2002, 214, 13–17. [Google Scholar] [CrossRef]
- Cole, D.; Snowden, K.; Cohen, N.; Smith, R. Detection of Cryptosporidium parvum in horses: Thresholds of acid-fast stain, immunofluorescence assay, and flow cytometry. J. Clin. Microbiol. 1999, 37, 457–460. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.; Hamady, M.; Knight, R. UniFrac-an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinform. 2006, 7, 371–385. [Google Scholar] [CrossRef] [Green Version]
- Westcott, S.L.; Schloss, P.D. OptiClust, an Improved Method for Assigning Amplicon-Based Sequence Data to Operational Taxonomic Units. mSphere 2017, 2, e00073-17. [Google Scholar] [CrossRef] [Green Version]
- Clarke, K.R. Non-parametric Multivariate Analyses of Changes in Community Structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Fisher, R.A. The use of multiple measurements in taxonomic problems. Ann. Eugen. 1936, 7, 179–188. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepš, J.; Šmilauer, P. Multivariate Analysis of Ecological Data Using CANOCO; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Legendre, P.; Anderson, M.J. Distance-based redundancy analysis: Testing multispecies responses in multifactorial ecological experiments. Ecol. Monogr. 1999, 69, 1–24. [Google Scholar] [CrossRef]
- Tichy, L.; Chytry, M. Statistical determination of diagnostic species for site groups of unequal size. J. Veg. Sci. 2006, 17, 809–818. [Google Scholar] [CrossRef]
- Griffiths, J.K.; Theodos, C.; Paris, M.; Tzipori, S. The gamma interferon gene knockout mouse: A highly sensitive model for evaluation of therapeutic agents against Cryptosporidium parvum. J. Clin. Microbiol. 1998, 36, 2503–2508. [Google Scholar] [CrossRef] [Green Version]
- Ungar, B.L.; Burris, J.A.; Quinn, C.A.; Finkelman, F.D. New mouse models for chronic Cryptosporidium infection in immunodeficient hosts. Infect. Immun. 1990, 58, 961–969. [Google Scholar] [CrossRef] [Green Version]
- Braak, C.T.; Šmilauer, P. CANOCO Reference Manual and CanoDraw for Windows User’S Guide: Software for Canonical Community Ordination (Version 4.5); Microcomputer Power: Ithaca, NY, USA, 2002. [Google Scholar]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 2016, 167, 1339–1353.e21. [Google Scholar] [CrossRef] [Green Version]
- Vasilescu, I.M.; Chifiriuc, M.C.; Pircalabioru, G.G.; Filip, R.; Bolocan, A.; Lazăr, V.; Diţu, L.M.; Bleotu, C. Gut Dysbiosis and Clostridioides difficile Infection in Neonates and Adults. Front. Microbiol. 2021, 12, 651081. [Google Scholar] [CrossRef]
- Winter, S.E.; Lopez, C.A.; Baumler, A.J. The dynamics of gut-associated microbial communities during inflammation. EMBO Rep. 2013, 14, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Gillis, C.C.; Hughes, E.R.; Spiga, L.; Winter, M.G.; Zhu, W.; Furtado de Carvalho, T.; Chanin, R.B.; Behrendt, C.L.; Hooper, L.V.; Santos, R.L.; et al. Dysbiosis-Associated Change in Host Metabolism Generates Lactate to Support Salmonella Growth. Cell Host Microbe 2018, 23, 54–64.e6. [Google Scholar] [CrossRef] [PubMed]
- Litvak, Y.; Byndloss, M.X.; Bäumler, A.J. Colonocyte metabolism shapes the gut microbiota. Science 2018, 362, eaat9076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ericsson, A.C.; Davis, J.W.; Spollen, W.; Bivens, N.; Givan, S.; Hagan, C.E.; McIntosh, M.; Franklin, C.L. Effects of vendor and genetic background on the composition of the fecal microbiota of inbred mice. PLoS ONE 2015, 10, e0116704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, L.L.; Svenson, K.L.; Mourino, A.J.; Michaud, M.; Fahey, J.R.; Waterman, L.; Vandegrift, K.L.; Adams, M.D. Shared and distinctive features of the gut microbiome of C57BL/6 mice from different vendors and production sites, and in response to a new vivarium. Lab Anim. 2021, 50, 185–195. [Google Scholar] [CrossRef]
- Koren, O.; Goodrich, J.K.; Cullender, T.C.; Spor, A.; Laitinen, K.; Backhed, H.K.; Gonzalez, A.; Werner, J.J.; Angenent, L.T.; Knight, R.; et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 2012, 150, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Capuco, A.; Urits, I.; Hasoon, J.; Chun, R.; Gerald, B.; Wang, J.K.; Ngo, A.L.; Simopoulos, T.; Kaye, A.D.; Colontonio, M.M.; et al. Gut Microbiome Dysbiosis and Depression: A Comprehensive Review. Curr. Pain Headache Rep. 2020, 24, 36. [Google Scholar] [CrossRef]
- Rogers, G.B.; Keating, D.J.; Young, R.L.; Wong, M.L.; Licinio, J.; Wesselingh, S. From gut dysbiosis to altered brain function and mental illness: Mechanisms and pathways. Mol. Psychiatry 2016, 21, 738–748. [Google Scholar] [CrossRef] [Green Version]
- Alegre, M.-L. Mouse microbiomes: Overlooked culprits of experimental variability. Genome Biol. 2019, 20, 108. [Google Scholar] [CrossRef]
- Hintze, K.J.; Cox, J.E.; Rompato, G.; Benninghoff, A.D.; Ward, R.E.; Broadbent, J.; Lefevre, M. Broad scope method for creating humanized animal models for animal health and disease research through antibiotic treatment and human fecal transfer. Gut Microbes 2014, 5, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Andoh, A.; Tsujikawa, T.; Fujiyama, Y. Role of dietary fiber and short-chain fatty acids in the colon. Curr. Pharm. Des. 2003, 9, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Marsman, K.E.; McBurney, M.I. Dietary fiber increases oxidative metabolism in colonocytes but not in distal small intestinal enterocytes isolated from rats. J. Nutr. 1995, 125, 273–282. [Google Scholar] [PubMed]
- Maslowski, K.M. Metabolism at the centre of the host–microbe relationship. Clin. Exp. Immunol. 2019, 197, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.M.; De Souza, R.; Kendall, C.W.; Emam, A.; Jenkins, D.J. Colonic health: Fermentation and short chain fatty acids. J. Clin. Gastroenterol. 2006, 40, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Alden, N.; Lee, K. Pathways and functions of gut microbiota metabolism impacting host physiology. Curr. Opin. Biotechnol. 2015, 36, 137–145. [Google Scholar] [CrossRef] [Green Version]
- VanDussen, K.L.; Funkhouser-Jones, L.J.; Akey, M.E.; Schaefer, D.A.; Ackman, K.; Riggs, M.W.; Stappenbeck, T.S.; Sibley, L.D. Neonatal mouse gut metabolites influence Cryptosporidium parvum infection in intestinal epithelial cells. MBio 2020, 11, e02582-20. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Finch, G.R.; Daniels, C.W.; Black, E.K.; Schaefer, F.W., 3rd; Belosevic, M. Dose response of Cryptosporidium parvum in outbred neonatal CD-1 mice. Appl. Environ. Microbiol. 1993, 59, 3661–3665. [Google Scholar] [CrossRef] [Green Version]
- Bonneau, R.H.; Sheridan, J.F.; Feng, N.; Glaser, R. Stress-induced suppression of herpes simplex virus (HSV)-specific cytotoxic T lymphocyte and natural killer cell activity and enhancement of acute pathogenesis following local HSV infection. Brain Behav. Immun. 1991, 5, 170–192. [Google Scholar] [CrossRef]
- Mead, J.R.; Arrowood, M.J.; Sidwell, R.W.; Healey, M.C. Chronic Cryptosporidium parvum infections in congenitally immunodeficient SCID and nude mice. J. Infect. Dis. 1991, 163, 1297–1304. [Google Scholar] [CrossRef]
- Tzipori, S. Cryptosporidiosis: Laboratory investigations and chemotherapy. Adv. Parasitol. 1998, 40, 187–221. [Google Scholar] [CrossRef]
- Cao, L.; Liu, X.; Lin, E.-J.D.; Wang, C.; Choi, E.Y.; Riban, V.; Lin, B.; During, M.J. Environmental and Genetic Activation of a Brain-Adipocyte BDNF/Leptin Axis Causes Cancer Remission and Inhibition. Cell 2010, 142, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Roberts, M.R.; Becker, S.M.; Podd, B.; Zhang, Y.; Chua, S.; Myers, M.; Duggal, P.; Houpt, E.R.; Petri, W. Leptin signaling in intestinal epithelium mediates resistance to enteric infection by Entamoeba histolytica. Mucosal Immunol. 2011, 4, 294–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, C.J. Effect of cage bedding on temperature regulation and metabolism of group-housed female mice. Comp. Med. 2004, 54, 63–68. [Google Scholar] [PubMed]
- Gavin, H.E.; Satchell, K.J.F. Surface hypothermia predicts murine mortality in the intragastric Vibrio vulnificus infection model. BMC Microbiol. 2017, 17, 136. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Martin, S.M.; Rubin, L.H.; McGee, K.M.; Shirk, E.N.; Queen, S.E.; Li, M.; Bullock, B.; Carlson, B.W.; Adams, R.J.; Gama, L. Psychosocial stress alters the immune response and results in higher viral load during acute simian immunodeficiency virus infection in a pigtailed macaque model of human immunodeficiency virus. J. Infect. Dis. 2021, 224, 2113–2121. [Google Scholar] [CrossRef]
- Yu, M.; Jia, H.; Zhou, C.; Yang, Y.; Zhao, Y.; Yang, M.; Zou, Z. Variations in gut microbiota and fecal metabolic phenotype associated with depression by 16S rRNA gene sequencing and LC/MS-based metabolomics. J. Pharm. Biomed. Anal. 2017, 138, 231–239. [Google Scholar] [CrossRef]
- Dillingham, R.; Lima, A.; Guerrant, R. Cryptosporidiosis: Epidemiology and impact. Microbes Infect. 2002, 4, 1059. [Google Scholar] [CrossRef]
- Lanternier, F.; Amazzough, K.; Favennec, L.; Mamzer-Bruneel, M.F.; Abdoul, H.; Tourret, J.; Decramer, S.; Zuber, J.; Scemla, A.; Legendre, C.; et al. Cryptosporidium spp. Infection in Solid Organ Transplantation: The Nationwide “TRANSCRYPTO” Study. Transplantation 2017, 101, 826–830. [Google Scholar]
- Costa, D.; Razakandrainibe, R.; Sautour, M.; Valot, S.; Basmaciyan, L.; Gargala, G.; Lemeteil, D.; Favennec, L.; Dalle, F. Human cryptosporidiosis in immunodeficient patients in France (2015–2017). Exp. Parasitol. 2018, 192, 108–112. [Google Scholar] [CrossRef]
- Ambroise-Thomas, P. Parasitic diseases and immunodeficiencies. Parasitology 2001, 122, S65–S71. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exp. | Mouse Strain | Gender | Caging | Total Mice | C. parvum Isolate | Oocyst Counts | 16S Sequence |
|---|---|---|---|---|---|---|---|
| 35 | CD-1 | female | 4/1/1/1/1/1/1/1/1 | 12 | MD | yes | yes |
| 37 | CD-1 | female | 4/4/1/1/1/1/1/1 | 14 | TU114 | indiv. caged only | yes |
| 38 | CD-1 | male | 4/4/1/1/1/1/1/1 | 14 | TU114 | yes | yes |
| 40 | B6J.C3-Sst1 | male | 4/4/1/1/1/1/1/1 | 14 | TU114 | yes | no |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Creasey, H.N.; Zhang, W.; Widmer, G. Effect of Caging on Cryptosporidium parvum Proliferation in Mice. Microorganisms 2022, 10, 1242. https://doi.org/10.3390/microorganisms10061242
Creasey HN, Zhang W, Widmer G. Effect of Caging on Cryptosporidium parvum Proliferation in Mice. Microorganisms. 2022; 10(6):1242. https://doi.org/10.3390/microorganisms10061242
Chicago/Turabian StyleCreasey, Hannah N., Wen Zhang, and Giovanni Widmer. 2022. "Effect of Caging on Cryptosporidium parvum Proliferation in Mice" Microorganisms 10, no. 6: 1242. https://doi.org/10.3390/microorganisms10061242
APA StyleCreasey, H. N., Zhang, W., & Widmer, G. (2022). Effect of Caging on Cryptosporidium parvum Proliferation in Mice. Microorganisms, 10(6), 1242. https://doi.org/10.3390/microorganisms10061242