
Role of Host Small GTPases in Apicomplexan Parasite Infection


Abstract
:1. Introduction
2. Manipulation of the Host Cell Cytoskeleton in Apicomplexan Parasite Infection
3. Roles of the Host Small GTPases in Intracellular Pathogen Infection
4. Host Small GTPases in Apicomplexan Infection: The Roles of Rho GTPases
5. Host Small GTPases in Apicomplexan Infections: The Roles of Rab GTPases
6. Host Small GTPases in Apicomplexan Infection: The Roles of Ras, Arf and Ran GTPases
7. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. World Malaria Report 2021; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Bigna, J.J.; Tochie, J.N.; Tounouga, D.N.; Bekolo, A.O.; Ymele, N.S.; Youda, E.L.; Sime, P.S.; Nansseu, J.R. Global, regional, and country seroprevalence of Toxoplasma gondii in pregnant women: A systematic review, modelling and meta-analysis. Sci. Rep. 2020, 10, 12102. [Google Scholar] [CrossRef] [PubMed]
- Basavaraju, A. Toxoplasmosis in HIV infection: An overview. Trop. Parasitol. 2016, 6, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Gerace, E.; Lo Presti, V.D.M.; Biondo, C. Infection: Epidemiology, Pathogenesis, and Differential Diagnosis. Eur. J. Microbiol. Immunol. 2019, 9, 119–123. [Google Scholar] [CrossRef]
- Gubbels, M.J.; Duraisingh, M.T. Evolution of apicomplexan secretory organelles. Int. J. Parasitol. 2012, 42, 1071–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adl, S.M.; Leander, B.S.; Simpson, A.G.; Archibald, J.M.; Anderson, O.R.; Bass, D.; Bowser, S.S.; Brugerolle, G.; Farmer, M.A.; Karpov, S.; et al. Diversity, nomenclature, and taxonomy of protists. Syst. Biol. 2007, 56, 684–689. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, A.; Soldati-Favre, D. Amino Acid Metabolism in Apicomplexan Parasites. Metabolites 2021, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Shunmugam, S.; Arnold, C.S.; Dass, S.; Katris, N.J.; Botté, C.Y. The flexibility of Apicomplexa parasites in lipid metabolism. PLoS Pathog. 2022, 18, e1010313. [Google Scholar] [CrossRef]
- Counihan, N.A.; Modak, J.K.; de Koning-Ward, T.F. How Malaria Parasites Acquire Nutrients From Their Host. Front. Cell Dev. Biol. 2021, 9, 649184. [Google Scholar] [CrossRef]
- Cardoso, R.; Nolasco, S.; Gonçalves, J.; Cortes, H.C.; Leitão, A.; Soares, H. Besnoitia besnoiti and Toxoplasma gondii: Two apicomplexan strategies to manipulate the host cell centrosome and Golgi apparatus. Parasitology 2014, 141, 1436–1454. [Google Scholar] [CrossRef]
- Cardoso, R.; Wang, J.; Müller, J.; Rupp, S.; Leitão, A.; Hemphill, A. Modulation of cis- and trans- Golgi and the Rab9A-GTPase during infection by Besnoitia besnoiti, Toxoplasma gondii and Neospora caninum. Exp. Parasitol. 2018, 187, 75–85. [Google Scholar] [CrossRef]
- De Niz, M.; Caldelari, R.; Kaiser, G.; Zuber, B.; Heo, W.D.; Heussler, V.T.; Agop-Nersesian, C. Hijacking of the host cell Golgi by Plasmodium berghei liver stage parasites. J. Cell Sci. 2021, 134, jcs252213. [Google Scholar] [CrossRef]
- Kellermann, M.; Scharte, F.; Hensel, M. Manipulation of Host Cell Organelles by Intracellular Pathogens. Int. J. Mol. Sci. 2021, 22, 6484. [Google Scholar] [CrossRef]
- Romano, J.D.; Sonda, S.; Bergbower, E.; Smith, M.E.; Coppens, I. Toxoplasma gondii salvages sphingolipids from the host Golgi through the rerouting of selected Rab vesicles to the parasitophorous vacuole. Mol. Biol. Cell 2013, 24, 1974–1995. [Google Scholar] [CrossRef] [Green Version]
- Romano, J.D.; Nolan, S.J.; Porter, C.; Ehrenman, K.; Hartman, E.J.; Hsia, R.C.; Coppens, I. The parasite Toxoplasma sequesters diverse Rab host vesicles within an intravacuolar network. J. Cell Biol. 2017, 216, 4235–4254. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Weiss, L.M.; Orlofsky, A. Coordinate control of host centrosome position, organelle distribution, and migratory response by Toxoplasma gondii via host mTORC2. J. Biol. Chem. 2010, 285, 15611–15618. [Google Scholar] [CrossRef] [Green Version]
- Coppens, I.; Dunn, J.D.; Romano, J.D.; Pypaert, M.; Zhang, H.; Boothroyd, J.C.; Joiner, K.A. Toxoplasma gondii sequesters lysosomes from mammalian hosts in the vacuolar space. Cell 2006, 125, 261–274. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, R.; Soares, H.; Hemphill, A.; Leitão, A. Apicomplexans pulling the strings: Manipulation of the host cell cytoskeleton dynamics. Parasitology 2016, 143, 957–970. [Google Scholar] [CrossRef] [Green Version]
- Plattner, F.; Soldati-Favre, D. Hijacking of host cellular functions by the Apicomplexa. Annu. Rev. Microbiol. 2008, 62, 471–487. [Google Scholar] [CrossRef]
- Colonne, P.M.; Winchell, C.G.; Voth, D.E. Hijacking Host Cell Highways: Manipulation of the Host Actin Cytoskeleton by Obligate Intracellular Bacterial Pathogens. Front. Cell. Infect. Microbiol. 2016, 6, 107. [Google Scholar] [CrossRef] [Green Version]
- Frénal, K.; Soldati-Favre, D. Role of the parasite and host cytoskeleton in apicomplexa parasitism. Cell Host. Microbe 2009, 5, 602–611. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, K.R.; Morrissette, N.S.; LaChapelle, S.; Blader, I.J. Host cell invasion by Toxoplasma gondii is temporally regulated by the host microtubule cytoskeleton. Eukaryot. Cell 2010, 9, 1680–1689. [Google Scholar] [CrossRef] [Green Version]
- Takemae, H.; Sugi, T.; Kobayashi, K.; Gong, H.; Ishiwa, A.; Recuenco, F.C.; Murakoshi, F.; Iwanaga, T.; Inomata, A.; Horimoto, T.; et al. Characterization of the interaction between Toxoplasma gondii rhoptry neck protein 4 and host cellular β-tubulin. Sci. Rep. 2013, 3, 3199. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, V.; Combe, A.; David, V.; Malmquist, N.A.; Delorme, V.; Leroy, C.; Blazquez, S.; Ménard, R.; Tardieux, I. Host cell entry by apicomplexa parasites requires actin polymerization in the host cell. Cell Host Microbe 2009, 5, 259–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhabra, E.S.; Higgs, H.N. The many faces of actin: Matching assembly factors with cellular structures. Nat. Cell Biol. 2007, 9, 1110–1121. [Google Scholar] [CrossRef] [PubMed]
- Delorme-Walker, V.; Abrivard, M.; Lagal, V.; Anderson, K.; Perazzi, A.; Gonzalez, V.; Page, C.; Chauvet, J.; Ochoa, W.; Volkmann, N.; et al. Toxofilin upregulates the host cortical actin cytoskeleton dynamics, facilitating Toxoplasma invasion. J. Cell Sci. 2012, 125, 4333–4342. [Google Scholar] [CrossRef] [Green Version]
- Perez-Cordon, G.; Nie, W.; Schmidt, D.; Tzipori, S.; Feng, H. Involvement of host calpain in the invasion of Cryptosporidium parvum. Microbes Infect. 2011, 13, 103–107. [Google Scholar] [CrossRef] [Green Version]
- Sinai, A.P.; Webster, P.; Joiner, K.A. Association of host cell endoplasmic reticulum and mitochondria with the Toxoplasma gondii parasitophorous vacuole membrane: A high affinity interaction. J. Cell Sci. 1997, 110 Pt 17, 2117–2128. [Google Scholar] [CrossRef]
- Pernas, L.; Adomako-Ankomah, Y.; Shastri, A.J.; Ewald, S.E.; Treeck, M.; Boyle, J.P.; Boothroyd, J.C. Toxoplasma effector MAF1 mediates recruitment of host mitochondria and impacts the host response. PLoS Biol. 2014, 12, e1001845. [Google Scholar] [CrossRef] [Green Version]
- Kelly, F.D.; Wei, B.M.; Cygan, A.M.; Parker, M.L.; Boulanger, M.J.; Boothroyd, J.C. MAF1b Binds the Host Cell MIB Complex To Mediate Mitochondrial Association. mSphere 2017, 2, e00183-17. [Google Scholar] [CrossRef] [Green Version]
- Coppens, I.; Romano, J.D. Sitting in the driver’s seat: Manipulation of mammalian cell Rab GTPase functions by apicomplexan parasites. Biol. Cell 2020, 112, 187–195. [Google Scholar] [CrossRef]
- Rug, M.; Cyrklaff, M.; Mikkonen, A.; Lemgruber, L.; Kuelzer, S.; Sanchez, C.P.; Thompson, J.; Hanssen, E.; O’Neill, M.; Langer, C.; et al. Export of virulence proteins by malaria-infected erythrocytes involves remodeling of host actin cytoskeleton. Blood 2014, 124, 3459–3468. [Google Scholar] [CrossRef] [Green Version]
- Millholland, M.G.; Mishra, S.; Dupont, C.D.; Love, M.S.; Patel, B.; Shilling, D.; Kazanietz, M.G.; Foskett, J.K.; Hunter, C.A.; Sinnis, P.; et al. A host GPCR signaling network required for the cytolysis of infected cells facilitates release of apicomplexan parasites. Cell Host Microbe 2013, 13, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Vetter, I.R.; Wittinghofer, A. The guanine nucleotide-binding switch in three dimensions. Science 2001, 294, 1299–1304. [Google Scholar] [CrossRef] [Green Version]
- Buchsbaum, R.J. Rho activation at a glance. J. Cell Sci. 2007, 120, 1149–1152. [Google Scholar] [CrossRef] [Green Version]
- Scheffzek, K.; Ahmadian, M.R. GTPase activating proteins: Structural and functional insights 18 years after discovery. Cell. Mol. Life Sci. CMLS 2005, 62, 3014–3038. [Google Scholar] [CrossRef]
- Song, S.; Cong, W.; Zhou, S.; Shi, Y.; Dai, W.; Zhang, H.; Wang, X.; He, B.; Zhang, Q. Small GTPases: Structure, biological function and its interaction with nanoparticles. Asian J. Pharm. Sci. 2019, 14, 30–39. [Google Scholar] [CrossRef]
- Hardt, W.D.; Chen, L.M.; Schuebel, K.E.; Bustelo, X.R.; Galán, J.E. S. typhimurium encodes an activator of Rho GTPases that induces membrane ruffling and nuclear responses in host cells. Cell 1998, 93, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Galán, J.E. A salmonella protein antagonizes Rac-1 and Cdc42 to mediate host-cell recovery after bacterial invasion. Nature 1999, 401, 293–297. [Google Scholar] [CrossRef]
- Stebbins, C.E.; Galán, J.E. Modulation of host signaling by a bacterial mimic: Structure of the Salmonella effector SptP bound to Rac1. Mol. Cell 2000, 6, 1449–1460. [Google Scholar] [CrossRef]
- Popoff, M.R. Bacterial factors exploit eukaryotic Rho GTPase signaling cascades to promote invasion and proliferation within their host. Small GTPases 2014, 5, e983863. [Google Scholar] [CrossRef] [Green Version]
- Barth, H.; Olenik, C.; Sehr, P.; Schmidt, G.; Aktories, K.; Meyer, D.K. Neosynthesis and activation of Rho by Escherichia coli cytotoxic necrotizing factor (CNF1) reverse cytopathic effects of ADP-ribosylated Rho. J. Biol. Chem. 1999, 274, 27407–27414. [Google Scholar] [CrossRef] [Green Version]
- Blumenthal, B.; Hoffmann, C.; Aktories, K.; Backert, S.; Schmidt, G. The cytotoxic necrotizing factors from Yersinia pseudotuberculosis and from Escherichia coli bind to different cellular receptors but take the same route to the cytosol. Infect. Immun. 2007, 75, 3344–3353. [Google Scholar] [CrossRef] [Green Version]
- Woolery, A.R.; Yu, X.; LaBaer, J.; Orth, K. AMPylation of Rho GTPases subverts multiple host signaling processes. J. Biol. Chem. 2014, 289, 32977–32988. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Sheedlo, M.J.; Yu, K.; Tan, Y.; Nakayasu, E.S.; Das, C.; Liu, X.; Luo, Z.Q. Ubiquitination independent of E1 and E2 enzymes by bacterial effectors. Nature 2016, 533, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Zamudio-Meza, H.; Castillo-Alvarez, A.; González-Bonilla, C.; Meza, I. Cross-talk between Rac1 and Cdc42 GTPases regulates formation of filopodia required for dengue virus type-2 entry into HMEC-1 cells. J. Gen. Virol. 2009, 90, 2902–2911. [Google Scholar] [CrossRef]
- Wang, J.L.; Zhang, J.L.; Chen, W.; Xu, X.F.; Gao, N.; Fan, D.Y.; An, J. Roles of small GTPase Rac1 in the regulation of actin cytoskeleton during dengue virus infection. PLoS Negl. Trop. Dis. 2010, 4, e809. [Google Scholar] [CrossRef] [Green Version]
- Pontow, S.E.; Heyden, N.V.; Wei, S.; Ratner, L. Actin cytoskeletal reorganizations and coreceptor-mediated activation of rac during human immunodeficiency virus-induced cell fusion. J. Virol. 2004, 78, 7138–7147. [Google Scholar] [CrossRef] [Green Version]
- Zoughlami, Y.; Voermans, C.; Brussen, K.; van Dort, K.A.; Kootstra, N.A.; Maussang, D.; Smit, M.J.; Hordijk, P.L.; van Hennik, P.B. Regulation of CXCR4 conformation by the small GTPase Rac1: Implications for HIV infection. Blood 2012, 119, 2024–2032. [Google Scholar] [CrossRef] [Green Version]
- Swaine, T.; Dittmar, M.T. CDC42 Use in Viral Cell Entry Processes by RNA Viruses. Viruses 2015, 7, 6526–6536. [Google Scholar] [CrossRef]
- Spearman, P. Viral interactions with host cell Rab GTPases. Small GTPases 2018, 9, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Belov, G.A.; Habbersett, C.; Franco, D.; Ehrenfeld, E. Activation of cellular Arf GTPases by poliovirus protein 3CD correlates with virus replication. J. Virol. 2007, 81, 9259–9267. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Gong, R.; Qu, J.; Zhou, Y.; Liu, W.; Chen, M.; Liu, Y.; Zhu, Y.; Wu, J. Activation of the Ras/Raf/MEK pathway facilitates hepatitis C virus replication via attenuation of the interferon-JAK-STAT pathway. J. Virol. 2012, 86, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Porter, F.W.; Bochkov, Y.A.; Albee, A.J.; Wiese, C.; Palmenberg, A.C. A picornavirus protein interacts with Ran-GTPase and disrupts nucleocytoplasmic transport. Proc. Natl. Acad. Sci. USA 2006, 103, 12417–12422. [Google Scholar] [CrossRef] [Green Version]
- Bonfim-Melo, A.; Ferreira, É.; Mortara, R.A. Rac1/WAVE2 and Cdc42/N-WASP Participation in Actin-Dependent Host Cell Invasion by Extracellular Amastigotes of. Front. Microbiol. 2018, 9, 360. [Google Scholar] [CrossRef]
- Teixeira, T.L.; Cruz, L.; Mortara, R.A.; Da Silva, C.V. Revealing Annexin A2 and ARF-6 enrollment during Trypanosoma cruzi extracellular amastigote-host cell interaction. Parasit. Vectors 2015, 8, 493. [Google Scholar] [CrossRef] [Green Version]
- Dutra, J.M.F.; Bonilha, V.L.; De Souza, W.; Carvalho, T.M.U. Role of small GTPases in Trypanosoma cruzi invasion in MDCK cell lines. Parasitol. Res. 2005, 96, 171–177. [Google Scholar] [CrossRef]
- Lodge, R.; Descoteaux, A. Phagocytosis of Leishmania donovani amastigotes is Rac1 dependent and occurs in the absence of NADPH oxidase activation. Eur. J. Immunol. 2006, 36, 2735–2744. [Google Scholar] [CrossRef]
- Lerm, M.; Holm, A.; Seiron, A.; Särndahl, E.; Magnusson, K.E.; Rasmusson, B. Leishmania donovani requires functional Cdc42 and Rac1 to prevent phagosomal maturation. Infect. Immun. 2006, 74, 2613–2618. [Google Scholar] [CrossRef] [Green Version]
- Mitin, N.; Roberts, P.J.; Chenette, E.J.; Der, C.J. Posttranslational lipid modification of Rho family small GTPases. Methods Mol. Biol. 2012, 827, 87–95. [Google Scholar] [CrossRef]
- Wennerberg, K.; Der, C.J. Rho-family GTPases: It’s not only Rac and Rho (and I like it). J. Cell Sci. 2004, 117, 1301–1312. [Google Scholar] [CrossRef] [Green Version]
- Billker, O.; Popp, A.; Brinkmann, V.; Wenig, G.; Schneider, J.; Caron, E.; Meyer, T.F. Distinct mechanisms of internalization of Neisseria gonorrhoeae by members of the CEACAM receptor family involving Rac1- and Cdc42-dependent and -independent pathways. EMBO J. 2002, 21, 560–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehm, M.; Krause-Gruszczynska, M.; Rohde, M.; Tegtmeyer, N.; Takahashi, S.; Oyarzabal, O.A.; Backert, S. Major host factors involved in epithelial cell invasion of Campylobacter jejuni: Role of fibronectin, integrin beta1, FAK, Tiam-1, and DOCK180 in activating Rho GTPase Rac1. Front. Cell Infect. Microbiol. 2011, 1, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, C.; Nans, A.; Boucrot, E.; Hayward, R.D. Chlamydia exploits filopodial capture and a macropinocytosis-like pathway for host cell entry. PLoS Pathog. 2018, 14, e1007051. [Google Scholar] [CrossRef]
- Friebel, A.; Ilchmann, H.; Aepfelbacher, M.; Ehrbar, K.; Machleidt, W.; Hardt, W.D. SopE and SopE2 from Salmonella typhimurium activate different sets of RhoGTPases of the host cell. J. Biol. Chem. 2001, 276, 34035–34040. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; White, C.D.; Li, Z.; Sacks, D.B. Salmonella enterica serotype Typhimurium usurps the scaffold protein IQGAP1 to manipulate Rac1 and MAPK signalling. Biochem. J. 2011, 440, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Krause-Gruszczynska, M.; Rohde, M.; Hartig, R.; Genth, H.; Schmidt, G.; Keo, T.; König, W.; Miller, W.G.; Konkel, M.E.; Backert, S. Role of the small Rho GTPases Rac1 and Cdc42 in host cell invasion of Campylobacter jejuni. Cell. Microbiol. 2007, 9, 2431–2444. [Google Scholar] [CrossRef]
- Maruvada, R.; Zhu, L.; Pearce, D.; Zheng, Y.; Perfect, J.; Kwon-Chung, K.J.; Kim, K.S. Cryptococcus neoformans phospholipase B1 activates host cell Rac1 for traversal across the blood-brain barrier. Cell. Microbiol. 2012, 14, 1544–1553. [Google Scholar] [CrossRef] [Green Version]
- Prehna, G.; Ivanov, M.I.; Bliska, J.B.; Stebbins, C.E. Yersinia virulence depends on mimicry of host Rho-family nucleotide dissociation inhibitors. Cell 2006, 126, 869–880. [Google Scholar] [CrossRef] [Green Version]
- Burnham, C.A.; Shokoples, S.E.; Tyrrell, G.J. Rac1, RhoA, and Cdc42 participate in HeLa cell invasion by group B streptococcus. FEMS Microbiol. Lett. 2007, 272, 8–14. [Google Scholar] [CrossRef]
- Doye, A.; Mettouchi, A.; Bossis, G.; Clément, R.; Buisson-Touati, C.; Flatau, G.; Gagnoux, L.; Piechaczyk, M.; Boquet, P.; Lemichez, E. CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell 2002, 111, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Paone, S.; D’Alessandro, S.; Parapini, S.; Celani, F.; Tirelli, V.; Pourshaban, M.; Olivieri, A. Characterization of the erythrocyte GTPase Rac1 in relation to Plasmodium falciparum invasion. Sci. Rep. 2020, 10, 22054. [Google Scholar] [CrossRef]
- Parapini, S.; Paone, S.; Erba, E.; Cavicchini, L.; Pourshaban, M.; Celani, F.; Contini, A.; D’Alessandro, S.; Olivieri, A. In vitro antimalarial activity of inhibitors of the human GTPase Rac1. Antimicrob. Agents Chemother. 2021, 66, e01498-21. [Google Scholar] [CrossRef] [PubMed]
- Millholland, M.G.; Chandramohanadas, R.; Pizzarro, A.; Wehr, A.; Shi, H.; Darling, C.; Lim, C.T.; Greenbaum, D.C. The malaria parasite progressively dismantles the host erythrocyte cytoskeleton for efficient egress. Mol. Cell. Proteom. 2011, 10, M111.010678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messina, V.; Loizzo, S.; Travaglione, S.; Bertuccini, L.; Condello, M.; Superti, F.; Guidotti, M.; Alano, P.; Silvestrini, F.; Fiorentini, C. The bacterial protein CNF1 as a new strategy against Plasmodium falciparum cytoadherence. PLoS ONE 2019, 14, e0213529. [Google Scholar] [CrossRef]
- Na, R.-H.; Zhu, G.-H.; Luo, J.-X.; Meng, X.-J.; Cui, L.; Peng, H.-J.; Chen, X.-g.; Gomez-Cambronero, J. Enzymatically active Rho and Rac small-GTPases are involved in the establishment of the vacuolar membrane after Toxoplasma gondii invasion of host cells. BMC Microbiol. 2013, 13, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Zhou, L.; Wu, S.; Li, D.; Deng, S.; Peng, H. Host cell Rac1 GTPase facilitates Toxoplasma gondii invasion. Sci. China Life Sci. 2020, 63, 610–612. [Google Scholar] [CrossRef]
- Chen, X.M.; Huang, B.Q.; Splinter, P.L.; Orth, J.D.; Billadeau, D.D.; McNiven, M.A.; LaRusso, N.F. Cdc42 and the actin-related protein/neural Wiskott-Aldrich syndrome protein network mediate cellular invasion by Cryptosporidium parvum. Infect. Immun. 2004, 72, 3011–3021. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.M.; Splinter, P.L.; Tietz, P.S.; Huang, B.Q.; Billadeau, D.D.; LaRusso, N.F. Phosphatidylinositol 3-kinase and frabin mediate Cryptosporidium parvum cellular invasion via activation of Cdc42. J. Biol. Chem. 2004, 279, 31671–31678. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.; Baumgartner, M. Filopodia and membrane blebs drive efficient matrix invasion of macrophages transformed by the intracellular parasite Theileria annulata. PLoS ONE 2013, 8, e75577. [Google Scholar] [CrossRef] [Green Version]
- Schmeck, B.; Beermann, W.; van Laak, V.; Opitz, B.; Hocke, A.C.; Meixenberger, K.; Eitel, J.; Chakraborty, T.; Schmidt, G.; Barth, H.; et al. Listeria monocytogenes induced Rac1-dependent signal transduction in endothelial cells. Biochem. Pharmacol. 2006, 72, 1367–1374. [Google Scholar] [CrossRef]
- Agarwal, V.; Hammerschmidt, S. Cdc42 and the phosphatidylinositol 3-kinase-Akt pathway are essential for PspC-mediated internalization of pneumococci by respiratory epithelial cells. J. Biol. Chem. 2009, 284, 19427–19436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Liu, Y.; You, Q.; Yang, E.; Liu, B.; Wang, H.; Xu, S.; Nawaz, W.; Chen, D.; Wu, Z. NSC23766 and Ehop016 Suppress Herpes Simplex Virus-1 Replication by Inhibiting Rac1 Activity. Biol. Pharm. Bull. 2021, 44, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Dierkes, R.; Warnking, K.; Liedmann, S.; Seyer, R.; Ludwig, S.; Ehrhardt, C. The Rac1 inhibitor NSC23766 exerts anti-influenza virus properties by affecting the viral polymerase complex activity. PLoS ONE 2014, 9, e88520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodge, R.; Descoteaux, A. Leishmania donovani promastigotes induce periphagosomal F-actin accumulation through retention of the GTPase Cdc42. Cell Microbiol. 2005, 7, 1647–1658. [Google Scholar] [CrossRef]
- Morehead, J.; Coppens, I.; Andrews, N.W. Opsonization modulates Rac-1 activation during cell entry by Leishmania amazonensis. Infect. Immun. 2002, 70, 4571–4580. [Google Scholar] [CrossRef] [Green Version]
- Taoufiq, Z.; Gay, F.; Balvanyos, J.; Ciceron, L.; Tefit, M.; Lechat, P.; Mazier, D. Rho kinase inhibition in severe malaria: Thwarting parasite-induced collateral damage to endothelia. J. Infect. Dis. 2008, 197, 1062–1073. [Google Scholar] [CrossRef] [Green Version]
- Seixas, E.; Ramalho, J.S.; Mota, L.J.; Barral, D.C.; Seabra, M.C. Bacteria and protozoa differentially modulate the expression of Rab proteins. PLoS ONE 2012, 7, e39858. [Google Scholar] [CrossRef] [Green Version]
- Seixas, E.; Escrevente, C.; Seabra, M.C.; Barral, D.C. Rab GTPase regulation of bacteria and protozoa phagocytosis occurs through the modulation of phagocytic receptor surface expression. Sci. Rep. 2018, 8, 12998. [Google Scholar] [CrossRef]
- Ólafsson, E.B.; Barragan, A. The unicellular eukaryotic parasite Toxoplasma gondii hijacks the migration machinery of mononuclear phagocytes to promote its dissemination. Biol. Cell 2020, 112, 239–250. [Google Scholar] [CrossRef]
- da Silva, C.V.; da Silva, E.A.; Cruz, M.C.; Chavrier, P.; Mortara, R.A. ARF6, PI3-kinase and host cell actin cytoskeleton in Toxoplasma gondii cell invasion. Biochem. Biophys. Res. Commun. 2009, 378, 656–661. [Google Scholar] [CrossRef]
- Huber, S.; Bär, A.; Epp, S.; Schmuckli-Maurer, J.; Eberhard, N.; Humbel, B.M.; Hemphill, A.; Woods, K. Recruitment of Host Nuclear Pore Components to the Vicinity of Theileria Schizonts. Msphere 2020, 5, e00709-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Dickerson, J.B.; Guo, F.; Zheng, J.; Zheng, Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 7618–7623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemichez, E.; Aktories, K. Hijacking of Rho GTPases during bacterial infection. Exp. Cell Res. 2013, 319, 2329–2336. [Google Scholar] [CrossRef] [PubMed]
- Shutes, A.; Onesto, C.; Picard, V.; Leblond, B.; Schweighoffer, F.; Der, C.J. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J. Biol. Chem. 2007, 282, 35666–35678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardama, G.A.; Comin, M.J.; Hornos, L.; Gonzalez, N.; Defelipe, L.; Turjanski, A.G.; Alonso, D.F.; Gomez, D.E.; Menna, P.L. Preclinical development of novel Rac1-GEF signaling inhibitors using a rational design approach in highly aggressive breast cancer cell lines. Anticancer Agents Med. Chem. 2014, 14, 840–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalfa, T.A.; Pushkaran, S.; Mohandas, N.; Hartwig, J.H.; Fowler, V.M.; Johnson, J.F.; Joiner, C.H.; Williams, D.A.; Zheng, Y. Rac GTPases regulate the morphology and deformability of the erythrocyte cytoskeleton. Blood 2006, 108, 3637–3645. [Google Scholar] [CrossRef] [Green Version]
- López-Gómez, A.; Cano, V.; Moranta, D.; Morey, P.; García del Portillo, F.; Bengoechea, J.A.; Garmendia, J. Host cell kinases, α5 and β1 integrins, and Rac1 signalling on the microtubule cytoskeleton are important for non-typable Haemophilus influenzae invasion of respiratory epithelial cells. Microbiology 2012, 158, 2384–2398. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.M.; Keithly, J.S.; Paya, C.V.; LaRusso, N.F. Cryptosporidiosis. N. Engl. J. Med. 2002, 346, 1723–1731. [Google Scholar] [CrossRef]
- Clark, D.P. New insights into human cryptosporidiosis. Clin. Microbiol. Rev. 1999, 12, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Dobbelaere, D.; Heussler, V. Transformation of leukocytes by Theileria parva and T. annulata. Annu. Rev. Microbiol. 1999, 53, 1–42. [Google Scholar] [CrossRef]
- McGuire, K.; Manuja, A.; Russell, G.C.; Springbett, A.; Craigmile, S.C.; Nichani, A.K.; Malhotra, D.V.; Glass, E.J. Quantitative analysis of pro-inflammatory cytokine mRNA expression in Theileria annulata-infected cell lines derived from resistant and susceptible cattle. Vet. Immunol. Immunopathol. 2004, 99, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Haubert, D.; Gharib, N.; Rivero, F.; Wiegmann, K.; Hösel, M.; Krönke, M.; Kashkar, H. PtdIns(4,5)P-restricted plasma membrane localization of FAN is involved in TNF-induced actin reorganization. EMBO J. 2007, 26, 3308–3321. [Google Scholar] [CrossRef]
- Lemichez, E.; Flatau, G.; Bruzzone, M.; Boquet, P.; Gauthier, M. Molecular localization of the Escherichia coli cytotoxic necrotizing factor CNF1 cell-binding and catalytic domains. Mol. Microbiol. 1997, 24, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Yeo, J.C.; Wall, A.A.; Luo, L.; Stow, J.L. Sequential recruitment of Rab GTPases during early stages of phagocytosis. Cell. Logist. 2016, 6, e1140615. [Google Scholar] [CrossRef] [Green Version]
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef] [Green Version]
- Donaldson, J.G.; Jackson, C.L. ARF family G proteins and their regulators: Roles in membrane transport, development and disease. Nat. Rev. Mol. Cell Biol. 2011, 12, 362–375. [Google Scholar] [CrossRef]
- Sztul, E.; Chen, P.W.; Casanova, J.E.; Cherfils, J.; Dacks, J.B.; Lambright, D.G.; Lee, F.S.; Randazzo, P.A.; Santy, L.C.; Schürmann, A.; et al. ARF GTPases and their GEFs and GAPs: Concepts and challenges. Mol. Biol. Cell 2019, 30, 1249–1271. [Google Scholar] [CrossRef]
- Donaldson, J.G. Multiple roles for Arf6: Sorting, structuring, and signaling at the plasma membrane. J. Biol. Chem. 2003, 278, 41573–41576. [Google Scholar] [CrossRef] [Green Version]
- Boudhraa, Z.; Carmona, E.; Provencher, D.; Mes-Masson, A.M. Ran GTPase: A Key Player in Tumor Progression and Metastasis. Front. Cell Dev. Biol. 2020, 8, 345. [Google Scholar] [CrossRef]
- Kessel, R.G. Annulate lamellae: A last frontier in cellular organelles. Int. Rev. Cytol. 1992, 133, 43–120. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.; Karagenc, T.; Ritler, D.; Rottenberg, S.; Woods, K. Identification and characterisation of a Theileria annulata proline-rich microtubule and SH3 domain-interacting protein (TaMISHIP) that forms a complex with CLASP1, EB1, and CD2AP at the schizont surface. Cell. Microbiol. 2018, 20, e12838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antony, H.A.; Parija, S.C. Antimalarial drug resistance: An overview. Trop. Parasitol. 2016, 6, 30–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairhurst, R.M.; Dondorp, A.M. Artemisinin-Resistant Plasmodium falciparum Malaria. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montazeri, M.; Mehrzadi, S.; Sharif, M.; Sarvi, S.; Tanzifi, A.; Aghayan, S.A.; Daryani, A. Drug Resistance in. Front. Microbiol. 2018, 9, 2587. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Yin, J.; Cuny, G. Current status and challenges in drug discovery against the globally important zoonotic cryptosporidiosis. Anim. Dis. 2021, 1, 3. [Google Scholar] [CrossRef]
- Prudêncio, M.; Mota, M.M. Targeting host factors to circumvent anti-malarial drug resistance. Curr. Pharm. Des. 2013, 19, 290–299. [Google Scholar] [CrossRef]
- Chiang, C.Y.; Uzoma, I.; Moore, R.T.; Gilbert, M.; Duplantier, A.J.; Panchal, R.G. Mitigating the Impact of Antibacterial Drug Resistance through Host-Directed Therapies: Current Progress, Outlook, and Challenges. MBio 2018, 9, e01932-17. [Google Scholar] [CrossRef] [Green Version]
- Stanley, S.A.; Barczak, A.K.; Silvis, M.R.; Luo, S.S.; Sogi, K.; Vokes, M.; Bray, M.A.; Carpenter, A.E.; Moore, C.B.; Siddiqi, N.; et al. Identification of host-targeted small molecules that restrict intracellular Mycobacterium tuberculosis growth. PLoS Pathog. 2014, 10, e1003946. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Chen, W.; Zhu, H.; Chen, Y.; Wan, X.; Yang, N.; Xu, S.; Yu, C.; Chen, L. Helicobacter pylori induces increased expression of the vitamin d receptor in immune responses. Helicobacter 2014, 19, 37–47. [Google Scholar] [CrossRef]
- Ottosen, S.; Parsley, T.B.; Yang, L.; Zeh, K.; van Doorn, L.J.; van der Veer, E.; Raney, A.K.; Hodges, M.R.; Patick, A.K. In vitro antiviral activity and preclinical and clinical resistance profile of miravirsen, a novel anti-hepatitis C virus therapeutic targeting the human factor miR-122. Antimicrob Agents Chemother. 2015, 59, 599–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Wispelaere, M.; LaCroix, A.J.; Yang, P.L. The small molecules AZD0530 and dasatinib inhibit dengue virus RNA replication via Fyn kinase. J. Virol. 2013, 87, 7367–7381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crouchet, E.; Wrensch, F.; Schuster, C.; Zeisel, M.B.; Baumert, T.F. Host-targeting therapies for hepatitis C virus infection: Current developments and future applications. Ther. Adv. Gastroenterol. 2018, 11, 1756284818759483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C.; et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 2005, 49, 4721–4732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glennon, E.K.K.; Dankwa, S.; Smith, J.D.; Kaushansky, A. Opportunities for Host-targeted Therapies for Malaria. Trends Parasitol. 2018, 34, 843–860. [Google Scholar] [CrossRef]
- Szewczyk-Golec, K.; Pawłowska, M.; Wesołowski, R.; Wróblewski, M.; Mila-Kierzenkowska, C. Oxidative Stress as a Possible Target in the Treatment of Toxoplasmosis: Perspectives and Ambiguities. Int. J. Mol. Sci. 2021, 22, 5705. [Google Scholar] [CrossRef]
- Brizuela, M.; Huang, H.M.; Smith, C.; Burgio, G.; Foote, S.J.; McMorran, B.J. Treatment of erythrocytes with the 2-cys peroxiredoxin inhibitor, Conoidin A, prevents the growth of Plasmodium falciparum and enhances parasite sensitivity to chloroquine. PLoS ONE 2014, 9, e92411. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.M.; Jerkovic, A.; Puy, H.; Winship, I.; Deybach, J.C.; Gouya, L.; van Dooren, G.; Goodman, C.D.; Sturm, A.; Manceau, H.; et al. Red cells from ferrochelatase-deficient erythropoietic protoporphyria patients are resistant to growth of malarial parasites. Blood 2015, 125, 534–541. [Google Scholar] [CrossRef]
- Hamie, M.; Najm, R.; Deleuze-Masquefa, C.; Bonnet, P.A.; Dubremetz, J.F.; El Sabban, M.; El Hajj, H. Imiquimod Targets Toxoplasmosis through Modulating Host Toll-Like Receptor-MyD88 Signaling. Front. Immunol. 2021, 12, 629917. [Google Scholar] [CrossRef]
- Castillo-Pichardo, L.; Humphries-Bickley, T.; De La Parra, C.; Forestier-Roman, I.; Martinez-Ferrer, M.; Hernandez, E.; Vlaar, C.; Ferrer-Acosta, Y.; Washington, A.V.; Cubano, L.A.; et al. The Rac Inhibitor EHop-016 Inhibits Mammary Tumor Growth and Metastasis in a Nude Mouse Model. Transl. Oncol. 2014, 7, 546–555. [Google Scholar] [CrossRef] [Green Version]
- Humphries-Bickley, T.; Castillo-Pichardo, L.; Corujo-Carro, F.; Duconge, J.; Hernandez-O’Farrill, E.; Vlaar, C.; Rodriguez-Orengo, J.F.; Cubano, L.; Dharmawardhane, S. Pharmacokinetics of Rac inhibitor EHop-016 in mice by ultra-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2015, 981–982, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto-Dominguez, N.; Parnell, C.; Teng, Y. Drugging the Small GTPase Pathways in Cancer Treatment: Promises and Challenges. Cells 2019, 8, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bid, H.K.; Roberts, R.D.; Manchanda, P.K.; Houghton, P.J. RAC1: An emerging therapeutic option for targeting cancer angiogenesis and metastasis. Mol. Cancer Ther. 2013, 12, 1925–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genetu Bayih, A.; Debnath, A.; Mitre, E.; Huston, C.D.; Laleu, B.; Leroy, D.; Blasco, B.; Campo, B.; Wells, T.N.C.; Willis, P.A.; et al. Susceptibility Testing of Medically Important Parasites. Clin. Microbiol. Rev. 2017, 30, 647–669. [Google Scholar] [CrossRef] [Green Version]
- Meneceur, P.; Bouldouyre, M.A.; Aubert, D.; Villena, I.; Menotti, J.; Sauvage, V.; Garin, J.F.; Derouin, F. In vitro susceptibility of various genotypic strains of Toxoplasma gondii to pyrimethamine, sulfadiazine, and atovaquone. Antimicrob. Agents Chemother. 2008, 52, 1269–1277. [Google Scholar] [CrossRef] [Green Version]
- Makler, M.T.; Ries, J.M.; Williams, J.A.; Bancroft, J.E.; Piper, R.C.; Gibbins, B.L.; Hinrichs, D.J. Parasite lactate dehydrogenase as an assay for Plasmodium falciparum drug sensitivity. Am. J. Trop. Med. Hyg. 1993, 48, 739–741. [Google Scholar] [CrossRef]
- Konreddy, A.K.; Rani, G.U.; Lee, K.; Choi, Y. Recent Drug-Repurposing-Driven Advances in the Discovery of Novel Antibiotics. Curr. Med. Chem. 2019, 26, 5363–5388. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, J.; Mohan, M.; Byrareddy, S.N. Drug Repurposing Approaches to Combating Viral Infections. J. Clin. Med. 2020, 9, 3777. [Google Scholar] [CrossRef]
- Tiede, I.; Fritz, G.; Strand, S.; Poppe, D.; Dvorsky, R.; Strand, D.; Lehr, H.A.; Wirtz, S.; Becker, C.; Atreya, R.; et al. CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J. Clin. Invest. 2003, 111, 1133–1145. [Google Scholar] [CrossRef] [Green Version]
- Oprea, T.I.; Sklar, L.A.; Agola, J.O.; Guo, Y.; Silberberg, M.; Roxby, J.; Vestling, A.; Romero, E.; Surviladze, Z.; Murray-Krezan, C.; et al. Novel Activities of Select NSAID R-Enantiomers against Rac1 and Cdc42 GTPases. PLoS ONE 2015, 10, e0142182. [Google Scholar] [CrossRef]
- Bobbala, D.; Koka, S.; Geiger, C.; Föller, M.; Huber, S.M.; Lang, F. Azathioprine favourably influences the course of malaria. Malar. J. 2009, 8, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
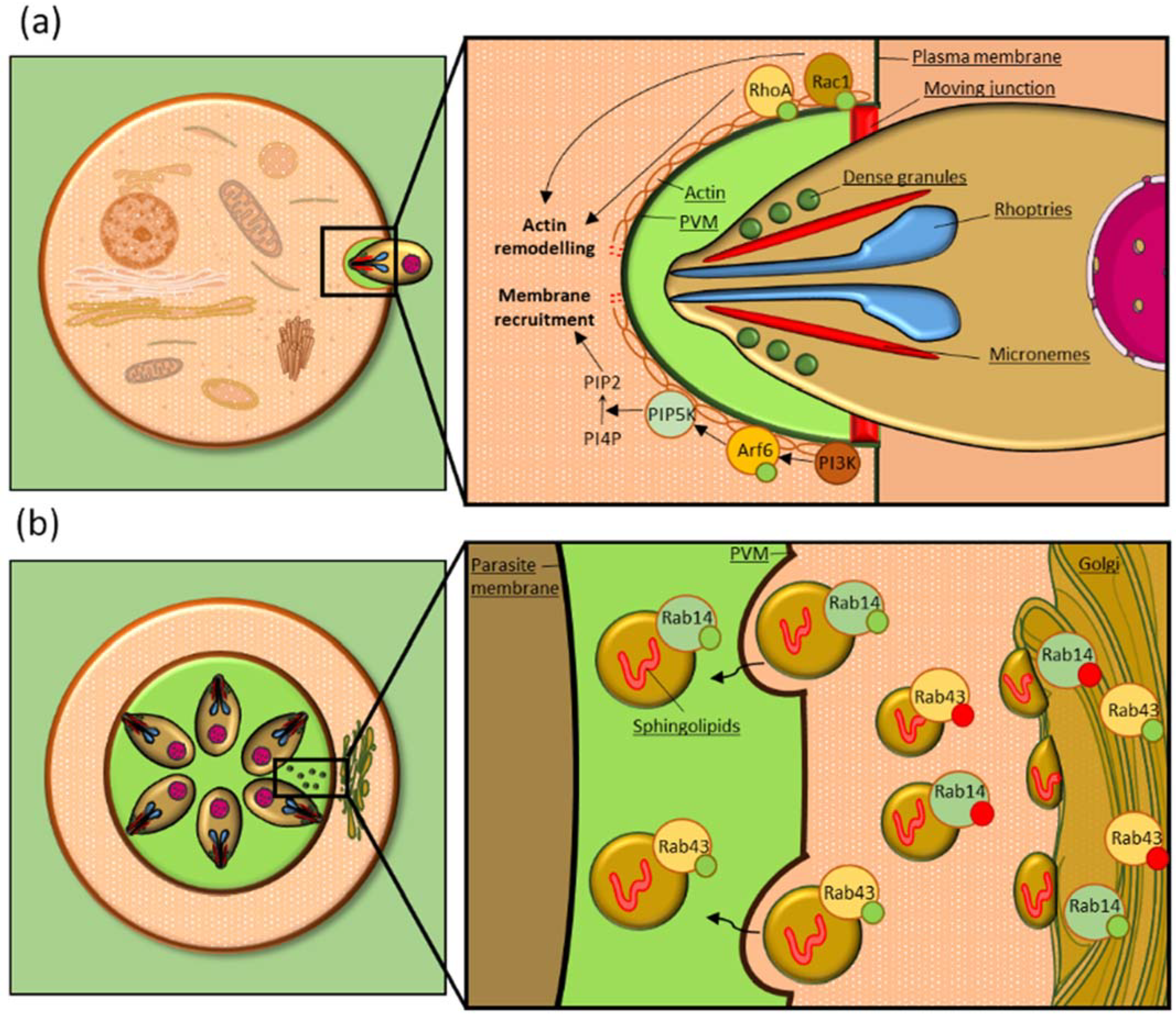

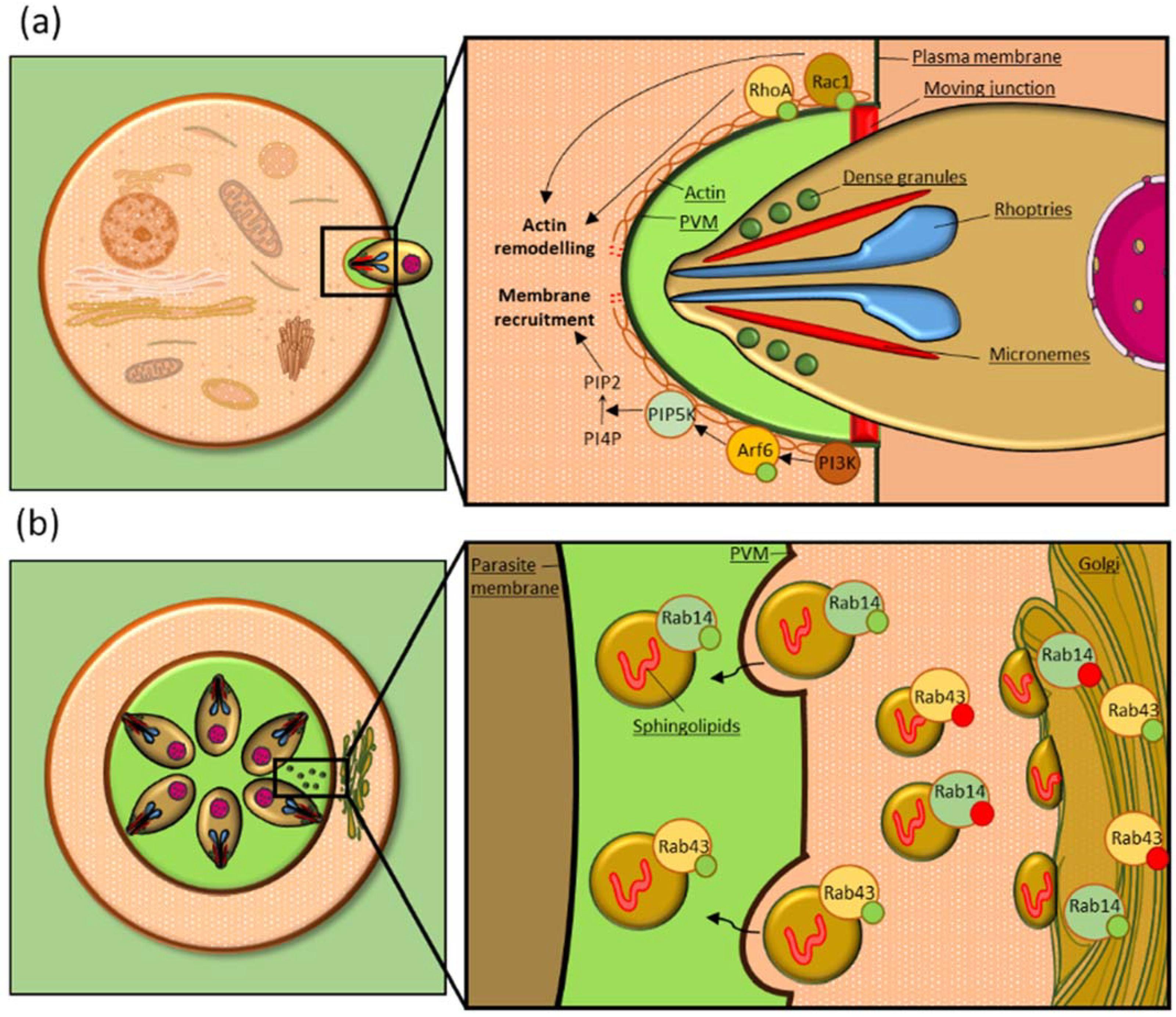{kind=link}
| Small GTPases Family | GTPase | Function | Organism |
|---|---|---|---|
| Rho | Rac1 | Invasion | P. falciparum [72,73] |
| Intracellular Growth | P. falciparum [72,73], T. gondii [77] | ||
| Egress | P. falciparum [74] | ||
| Cytoadherence | P. falciparum [75] | ||
| Cdc42 | Invasion | C. parvum [78,79] | |
| Cytoadherence | P. falciparum [75] | ||
| Infected cell motility | T. annulate [80] | ||
| RhoA | Invasion | T. gondii [76] | |
| Cytoadherence | P. falciparum [75,87] | ||
| Rab | Rab1 | Nutrients scavenging | T. gondii [15] |
| Rab11 | Nutrients scavenging | P. berghei [12] | |
| Rab14 | Nutrients scavenging | T. gondii [14] | |
| Immune evasion | P. berghei [88,89] | ||
| Rab43 | Nutrients scavenging | T. gondii [14] | |
| Ras | Ras | Infected cell motility | T. gondii [90] |
| Arf | Arf6 | Infected cell motility | T. gondii [91] |
| Ran | Ran | Nuclear delivery of parasite factors | T. annulate [92] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paone, S.; Olivieri, A. Role of Host Small GTPases in Apicomplexan Parasite Infection. Microorganisms 2022, 10, 1370. https://doi.org/10.3390/microorganisms10071370
Paone S, Olivieri A. Role of Host Small GTPases in Apicomplexan Parasite Infection. Microorganisms. 2022; 10(7):1370. https://doi.org/10.3390/microorganisms10071370
Chicago/Turabian StylePaone, Silvio, and Anna Olivieri. 2022. "Role of Host Small GTPases in Apicomplexan Parasite Infection" Microorganisms 10, no. 7: 1370. https://doi.org/10.3390/microorganisms10071370
APA StylePaone, S., & Olivieri, A. (2022). Role of Host Small GTPases in Apicomplexan Parasite Infection. Microorganisms, 10(7), 1370. https://doi.org/10.3390/microorganisms10071370