
Distribution of Core Root Microbiota of Tibetan Hulless Barley along an Altitudinal and Geographical Gradient in the Tibetan Plateau


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Analysis of Soil Chemical and Metabolite Properties
2.2. Extraction of DNA from Root Microbiome Samples
2.3. 16S rRNA Gene and ITS Region Bioinformatics and Phylogenetic Analyses
2.4. Diversity and Statistical Analyses
2.5. Random Forest Classification and Prediction
2.6. Phylogenetic Tree of Core Microbial Taxa
3. Results
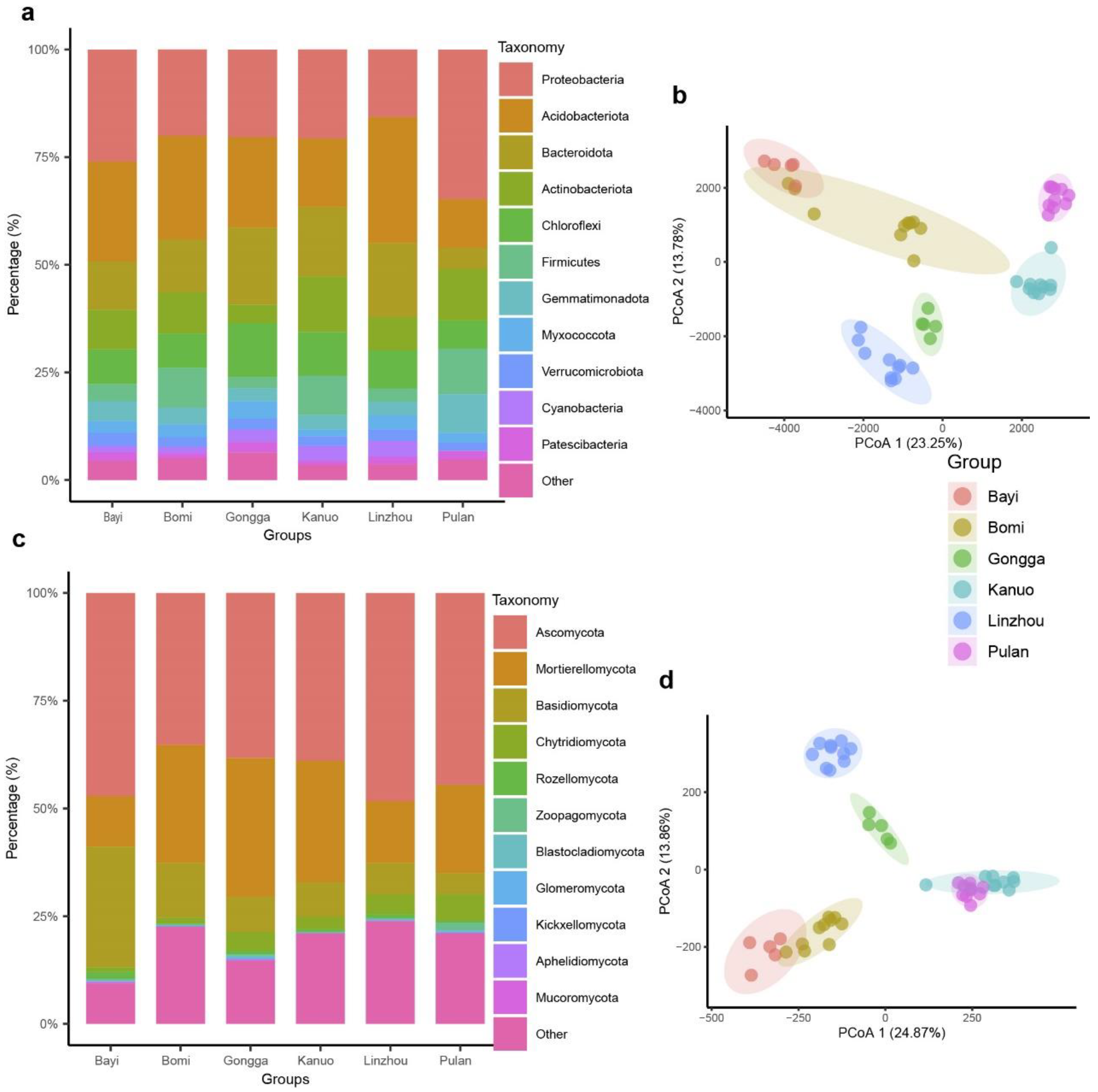
3.1. Rhizosphere Microbial Community Composition of Tibetan Hull-Less Barley
3.2. Rhizosphere Microbial Diversity and Community Structure of Tibetan Hull-Less Barley
3.3. Core and Enriched Rhizosphere Bacterial and Fungal ASVs
3.4. Environmental Factors Shaping Rhizosphere Microbiota and the Biomarkers Distinguishing Different Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Newman, C.; Newman, R.K. A brief history of barley foods. Cereal Foods World 2006, 51, 4–7. [Google Scholar] [CrossRef]
- Badr, A.; Rabey, H.E.; Effgen, S.; Ibrahim, H.; Pozzi, C.; Rohde, W.; Salamini, F. On the origin and domestication history of barley (Hordeum vulgare). Mol. Biol. Evol. 2000, 17, 499–510. [Google Scholar] [CrossRef]
- Zeng, X.; Guo, Y.; Xu, Q.; Mascher, M.; Guo, G.; Li, S.; Mao, L.; Liu, Q.; Xia, Z.; Zhou, J. Origin and evolution of qingke barley in Tibet. Nat. Commun. 2018, 9, 5433. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.H.; Dong, G.H.; Zhang, D.J.; Liu, X.Y.; Jia, X.; An, C.-B.; Ma, M.M.; Xie, Y.W.; Barton, L.; Ren, X. Agriculture facilitated permanent human occupation of the Tibetan Plateau after 3600 BP. Science 2015, 347, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Thompson, L.G.; Mosbrugger, V.; Zhang, F.; Ma, Y.; Luo, T.; Xu, B.; Yang, X.; Joswiak, D.R.; Wang, W.; et al. Third Pole Environment (TPE). Environ. Dev. 2012, 3, 52–64. [Google Scholar] [CrossRef]
- Gong, L.; Gong, L.; Zhang, Y. Intake of Tibetan Hulless Barley is Associated with a Reduced Risk of Metabolic Related Syndrome in Rats Fed High-Fat-Sucrose Diets. Nutrients 2014, 6, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.; Tsolakidou, M.-D.; Stringlis, I.A.; Pantelides, I.S. Rhizosphere Microbiome Recruited from a Suppressive Compost Improves Plant Fitness and Increases Protection against Vascular Wilt Pathogens of Tomato. Front. Plant Sci. 2017, 8, 2022. [Google Scholar] [CrossRef]
- Pii, Y.; Mimmo, T.; Tomasi, N.; Terzano, R.; Cesco, S.; Crecchio, C. Microbial interactions in the rhizosphere: Beneficial influences of plant growth-promoting rhizobacteria on nutrient acquisition process. A review. Biol. Fert. Soils 2015, 51, 403–415. [Google Scholar] [CrossRef]
- Edwards, J.; Johnson, C.; Santos-Medellin, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef]
- Dini-Andreote, F.; Raaijmakers, J.M. Engineering the Rhizosphere. Trends Plant Sci. 2016, 21, 266–278. [Google Scholar] [CrossRef]
- Philippot, L.; Raaijmakers, J.M.; Lemanceau, P.; van der Putten, W.H. Going back to the roots: The microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 2013, 11, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Bardgett, R.D.; Van Der Putten, W.H. Belowground biodiversity and ecosystem functioning. Nature 2014, 515, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, J.-T.; Wang, W.-H.; Tsui, C.K.M.; Cai, L. Changes in Bacterial and Fungal Microbiomes Associated with Tomatoes of Healthy and Infected by Fusarium oxysporum f. sp. lycopersici. Microbiol. Ecol. 2021, 81, 1004–1017. [Google Scholar] [CrossRef] [PubMed]
- Lareen, A.; Burton, F.; Schafer, P. Plant root-microbe communication in shaping root microbiomes. Plant Mol. Biol. 2016, 90, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Zhalnina, K.; Louie, K.B.; Hao, Z.; Mansoori, N.; da Rocha, U.N.; Shi, S.; Cho, H.; Karaoz, U.; Loqué, D.; Bowen, B.P.; et al. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat. Microbiol. 2018, 3, 470–480. [Google Scholar] [CrossRef]
- Durán, P.; Thiergart, T.; Garrido-Oter, R.; Agler, M.; Kemen, E.; Schulze-Lefert, P.; Hacquard, S. Microbial Interkingdom Interactions in Roots Promote Arabidopsis Survival. Cell 2018, 175, 973–983.e14. [Google Scholar] [CrossRef]
- Rahman, M.M.; Flory, E.; Koyro, H.W.; Abideen, Z.; Schikora, A.; Suarez, C.; Schnell, S.; Cardinale, M. Consistent associations with beneficial bacteria in the seed endosphere of barley (Hordeum vulgare L.). Syst. Appl. Microbiol. 2018, 41, 386–398. [Google Scholar] [CrossRef]
- Sapkota, R.; Knorr, K.; Jorgensen, L.N.; O’Hanlon, K.A.; Nicolaisen, M. Host genotype is an important determinant of the cereal phyllosphere mycobiome. New Phytol. 2015, 207, 1134–1144. [Google Scholar] [CrossRef]
- Guan, X.; Cheng, Z.; Li, Y.; Zhao, R.; Guo, Z.; Zhao, T.; Huang, L.; Qiu, C.; Chen, L.; Shi, W. Mixed Organic and Inorganic Amendments Enhance Soil Microbial Interactions and environmental stress resistance of Tibetan Barley on Plateau Farmland. Researchsquare 2022. [Google Scholar] [CrossRef]
- Kaur, T.; Devi, R.; Kumar, S.; Sheikh, I.; Kour, D.; Yadav, A.N. Microbial consortium with nitrogen fixing and mineral solubilizing attributes for growth of barley (Hordeum vulgare L.). Heliyon 2022, 8, e09326. [Google Scholar] [CrossRef]
- Zhao, Y.; Yao, Y.; Xu, H.; Xie, Z.; Guo, J.; Qi, Z.; Jiang, H. Soil metabolomics and bacterial functional traits revealed the responses of rhizosphere soil bacterial community to long-term continuous cropping of Tibetan barley. PeerJ 2022, 10, e13254. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.D.; Li, L.; Zhuo, G.; Xue, B. Characterizing Structure and Potential Function of Bacterial and Fungal Root Microbiota in Hulless Barley Cultivars. J. Soil Sci. Plant Nutr. 2019, 19, 420–429. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; Ver Loren van Themaat, E.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; Schmelzer, E.; et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.; Salas Gonzalez, I.; Mittelviefhaus, M.; Clingenpeel, S.; Herrera Paredes, S.; Miao, J.; Wang, K.; Devescovi, G.; Stillman, K.; Monteiro, F.; et al. Genomic features of bacterial adaptation to plants. Nat. Genet. 2017, 50, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, J.-T.; Zhang, Z.-F.; Li, W.; Chen, W.; Cai, L. Microbiota in the Rhizosphere and Seed of Rice From China, With Reference to Their Transmission and Biogeography. Front. Microbiol. 2020, 11, 995. [Google Scholar] [CrossRef]
- Klaedtke, S.; Jacques, M.A.; Raggi, L.; Preveaux, A.; Bonneau, S.; Negri, V.; Chable, V.; Barret, M. Terroir is a key driver of seed-associated microbial assemblages. Environ. Microbiol. 2016, 18, 1792–1804. [Google Scholar] [CrossRef]
- Abdelfattah, A.; Freilich, S.; Bartuv, R.; Zhimo, V.Y.; Kumar, A.; Biasi, A.; Salim, S.; Feygenberg, O.; Burchard, E.; Dardick, C.; et al. Global analysis of the apple fruit microbiome: Are all apples the same? Environ. Microbiol. 2021, 23, 6038–6055. [Google Scholar] [CrossRef]
- Fan, K.K.; Cardona, C.; Li, Y.T.; Shi, Y.; Xiang, X.J.; Shen, C.C.; Wang, H.F.; Gilbert, J.A.; Chu, H.Y. Rhizosphere-associated bacterial network structure and spatial distribution differ significantly from bulk soil in wheat crop fields. Soil Biol. Biochem. 2017, 113, 275–284. [Google Scholar] [CrossRef]
- Liu, X.; Yang, T.; Shi, Y.; Zhu, Y.; He, M.; Zhao, Y.; Adams, J.M.; Chu, H. Strong partitioning of soil bacterial community composition and co-occurrence networks along a small-scale elevational gradient on Zijin Mountain. Soil Ecol. Lett. 2021, 3, 290–302. [Google Scholar] [CrossRef]
- Qi, Z.; Zhou, X.; Tian, L.; Zhang, H.; Cai, L.; Tang, F. Distribution of mycotoxin-producing fungi across major rice production areas of China. Food Control 2022, 134, 108572. [Google Scholar] [CrossRef]
- Shamim, M.; Kumar, P.; Kumar, R.R.; Kumar, M.; Kumar, R.R.; Singh, K. Assessing Fungal Biodiversity Using Molecular Markers. In Molecular Markers in Mycology; Springer: Berlin/Heidelberg, Germany, 2017; pp. 305–333. [Google Scholar] [CrossRef]
- Edgar, R. Taxonomy annotation and guide tree errors in 16S rRNA databases. PeerJ 2018, 6, e5030. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Package ‘vegan’. Community Ecol. Package Version 2013, 2, 1–295. [Google Scholar]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef]
- Best, D.; Roberts, D. Algorithm AS 89: The upper tail probabilities of Spearman’s rho. J. R. Stat. Soc. Ser. C (Appl. Stat.) 1975, 24, 377–379. [Google Scholar] [CrossRef]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Y.-X.; Zhang, N.; Hu, B.; Jin, T.; Xu, H.; Qin, Y.; Yan, P.; Zhang, X.; Guo, X.; et al. NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotechnol. 2019, 37, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, J.; Carvalhais, L.C.; Percy, C.D.; Prakash Verma, J.; Schenk, P.M.; Singh, B.K. Evidence for the plant recruitment of beneficial microbes to suppress soil-borne pathogens. New Phytol. 2021, 229, 2873–2885. [Google Scholar] [CrossRef] [PubMed]
- Bahram, M.; Hildebrand, F.; Forslund, S.K.; Anderson, J.L.; Soudzilovskaia, N.A.; Bodegom, P.M.; Bengtsson-Palme, J.; Anslan, S.; Coelho, L.P.; Harend, H.; et al. Structure and function of the global topsoil microbiome. Nature 2018, 560, 233–237. [Google Scholar] [CrossRef]
- Edwards, J.A.; Santos-Medellin, C.M.; Liechty, Z.S.; Nguyen, B.; Lurie, E.; Eason, S.; Phillips, G.; Sundaresan, V. Compositional shifts in root-associated bacterial and archaeal microbiota track the plant life cycle in field-grown rice. PLoS Biol. 2018, 16, e2003862. [Google Scholar] [CrossRef]
- Jansson, J.K.; Hofmockel, K.S. Soil microbiomes and climate change. Nat. Rev. Microbiol. 2020, 18, 35–46. [Google Scholar] [CrossRef]
- Royden, L.H.; Burchfiel, B.C.; Hilst, R.D.v.d. The Geological Evolution of the Tibetan Plateau. Science 2008, 321, 1054–1058. [Google Scholar] [CrossRef]
- Giuliani, S.E.; Frank, A.M.; Corgliano, D.M.; Seifert, C.; Hauser, L.; Collart, F.R. Environment sensing and response mediated by ABC transporters. BMC Genom. 2011, 12 (Suppl. S1), S8. [Google Scholar] [CrossRef]
- Finkel, O.M.; Castrillo, G.; Herrera Paredes, S.; Salas Gonzalez, I.; Dangl, J.L. Understanding and exploiting plant beneficial microbes. Curr. Opin. Plant Biol. 2017, 38, 155–163. [Google Scholar] [CrossRef]
- Marasco, R.; Rolli, E.; Ettoumi, B.; Vigani, G.; Mapelli, F.; Borin, S.; Abou-Hadid, A.F.; El-Behairy, U.A.; Sorlini, C.; Cherif, A.; et al. A drought resistance-promoting microbiome is selected by root system under desert farming. PLoS ONE 2012, 7, e48479. [Google Scholar] [CrossRef]
- Walitang, D.I.; Kim, K.; Madhaiyan, M.; Kim, Y.K.; Kang, Y.; Sa, T. Characterizing endophytic competence and plant growth promotion of bacterial endophytes inhabiting the seed endosphere of Rice. BMC Microbiol. 2017, 17, 209. [Google Scholar] [CrossRef]
- Compant, S.; Samad, A.; Faist, H.; Sessitsch, A. A review on the plant microbiome: Ecology, functions, and emerging trends in microbial application. J. Adv. Res. 2019, 19, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shen, Z.; Li, C.; Kou, Y.; Wang, Y.; Tu, B.; Zhang, S.; Li, X. Stair-Step Pattern of Soil Bacterial Diversity Mainly Driven by pH and Vegetation Types Along the Elevational Gradients of Gongga Mountain, China. Front. Microbiol. 2018, 9, 569. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.-J.; Kong, H.G.; Choi, K.; Kwon, S.-K.; Song, J.Y.; Lee, J.; Lee, P.A.; Choi, S.Y.; Seo, M.; Lee, H.J.; et al. Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat. Biotechnol. 2018, 36, 1100–1109. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, B.; Zheng, S.; Zhang, X.; Wang, X.; Dong, W.; Xie, Q.; Wang, G.; Xiao, Y.; Chen, F.; et al. A phosphate starvation response-centered network regulates mycorrhizal symbiosis. Cell 2021, 184, 5527–5540.e18. [Google Scholar] [CrossRef]
- Lagier, J.C.; Khelaifia, S.; Alou, M.T.; Ndongo, S.; Dione, N.; Hugon, P.; Caputo, A.; Cadoret, F.; Traore, S.I.; Seck, E.H.; et al. Culture of previously uncultured members of the human gut microbiota by culturomics. Nat. Microbiol. 2016, 1, 16203. [Google Scholar] [CrossRef]
- Lewis, W.H.; Tahon, G.; Geesink, P.; Sousa, D.Z.; Ettema, T.J.G. Innovations to culturing the uncultured microbial majority. Nat. Rev. Microbiol. 2021, 19, 225–240. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, N.; Yue, X. Distribution of Core Root Microbiota of Tibetan Hulless Barley along an Altitudinal and Geographical Gradient in the Tibetan Plateau. Microorganisms 2022, 10, 1737. https://doi.org/10.3390/microorganisms10091737
Wei N, Yue X. Distribution of Core Root Microbiota of Tibetan Hulless Barley along an Altitudinal and Geographical Gradient in the Tibetan Plateau. Microorganisms. 2022; 10(9):1737. https://doi.org/10.3390/microorganisms10091737
Chicago/Turabian StyleWei, Na, and Xiaofeng Yue. 2022. "Distribution of Core Root Microbiota of Tibetan Hulless Barley along an Altitudinal and Geographical Gradient in the Tibetan Plateau" Microorganisms 10, no. 9: 1737. https://doi.org/10.3390/microorganisms10091737
APA StyleWei, N., & Yue, X. (2022). Distribution of Core Root Microbiota of Tibetan Hulless Barley along an Altitudinal and Geographical Gradient in the Tibetan Plateau. Microorganisms, 10(9), 1737. https://doi.org/10.3390/microorganisms10091737