
Morganella Phage Mecenats66 Utilizes an Evolutionarily Distinct Subtype of Headful Genome Packaging with a Preferred Packaging Initiation Site




Abstract
:1. Introduction
2. Materials and Methods
2.1. Host Bacteria and Phage Isolation and Propagation
2.2. Phage Concentration and Purification
2.3. Phage Sample Transmission Electron Microscopy and Virion Dimension Determination
2.4. Phage Genomic DNA Extraction, Whole Genome Sequencing and De Novo Assembly
2.5. Phage Genome De Novo Assembly and Functional Annotation
2.6. Taxonomic Identification of the Host
2.7. Phage Genome Termini/Packaging Strategy Prediction In Silico
2.8. Phage Genome Termini/Packaging Strategy Elucidation In Vitro
2.9. Representative TerL Sequence Diversity Evaluation
3. Results and Discussion
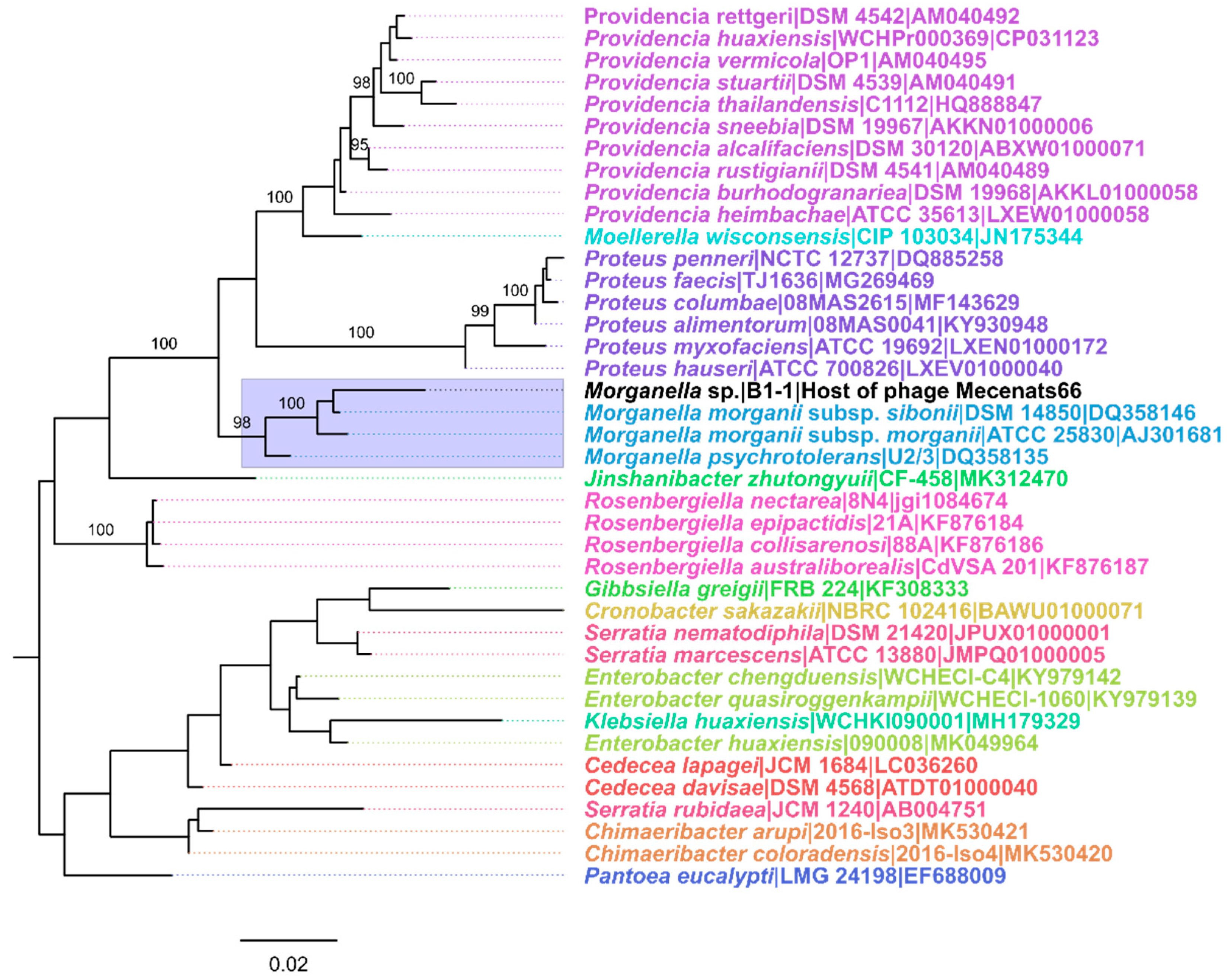
3.1. Host of Phage Mecenats66
3.2. Morganella Phage Mecenats66 Virion Morphology and Genome Overview
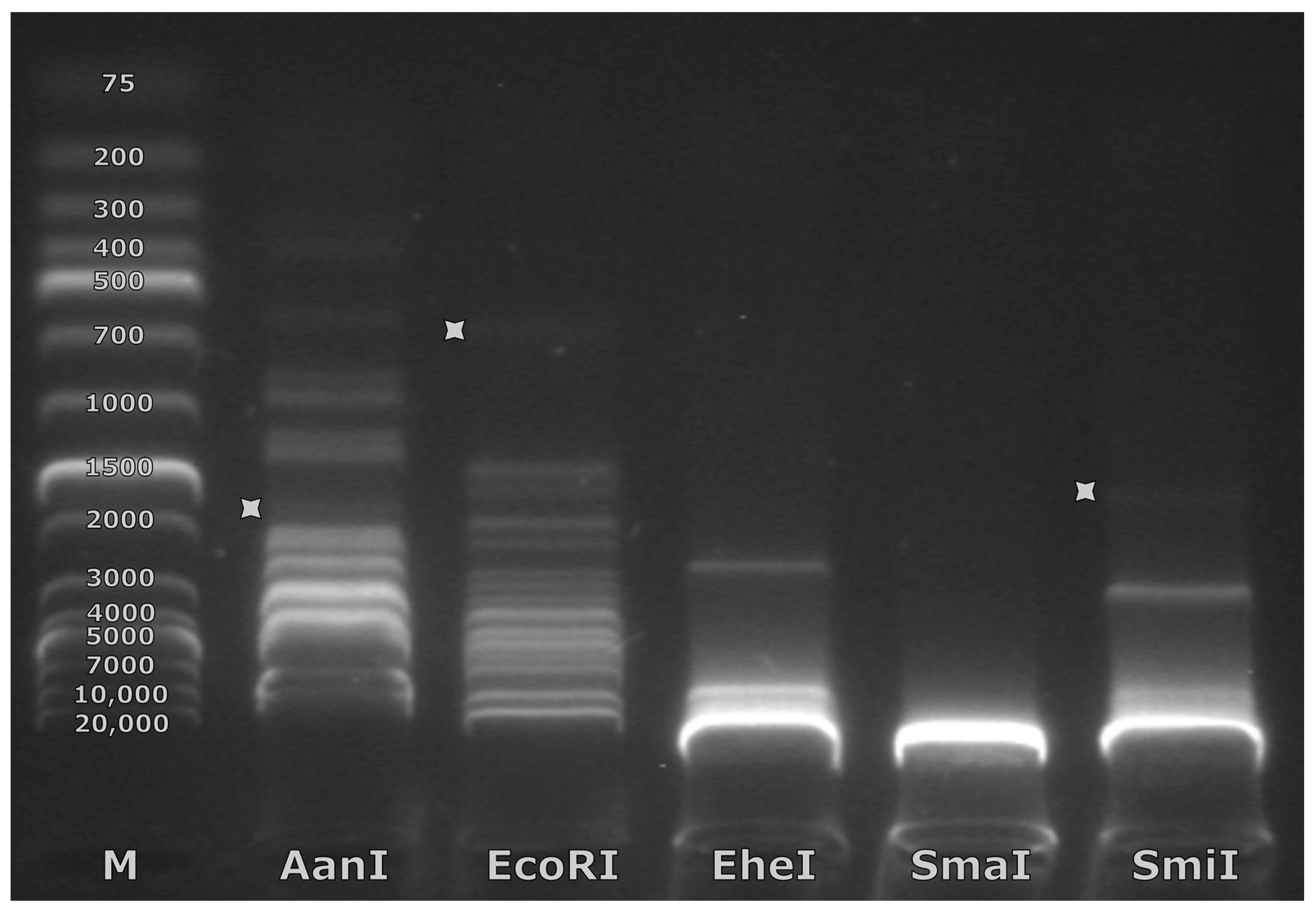
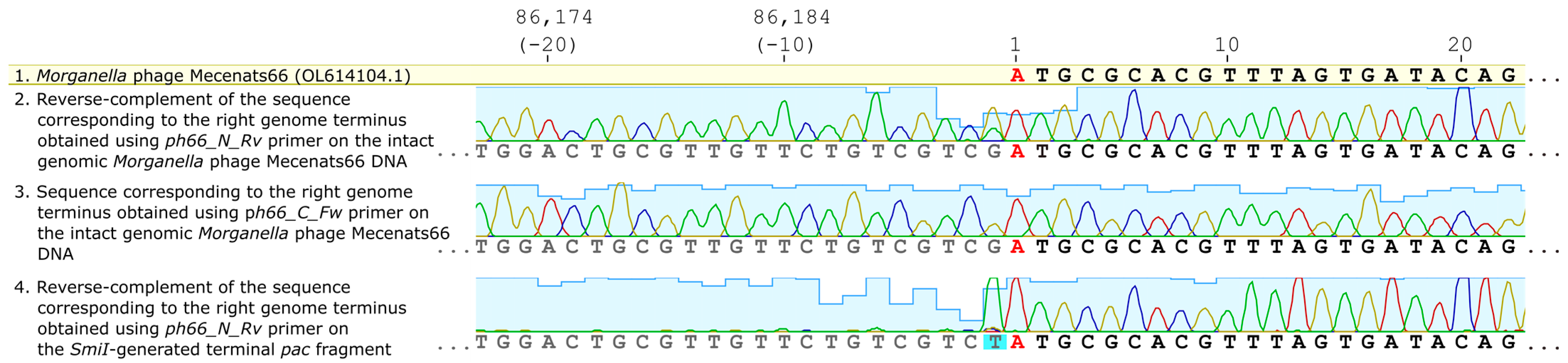
3.3. Exact Genome Termini and Genome Packaging Strategy Elucidation for Mecenats66
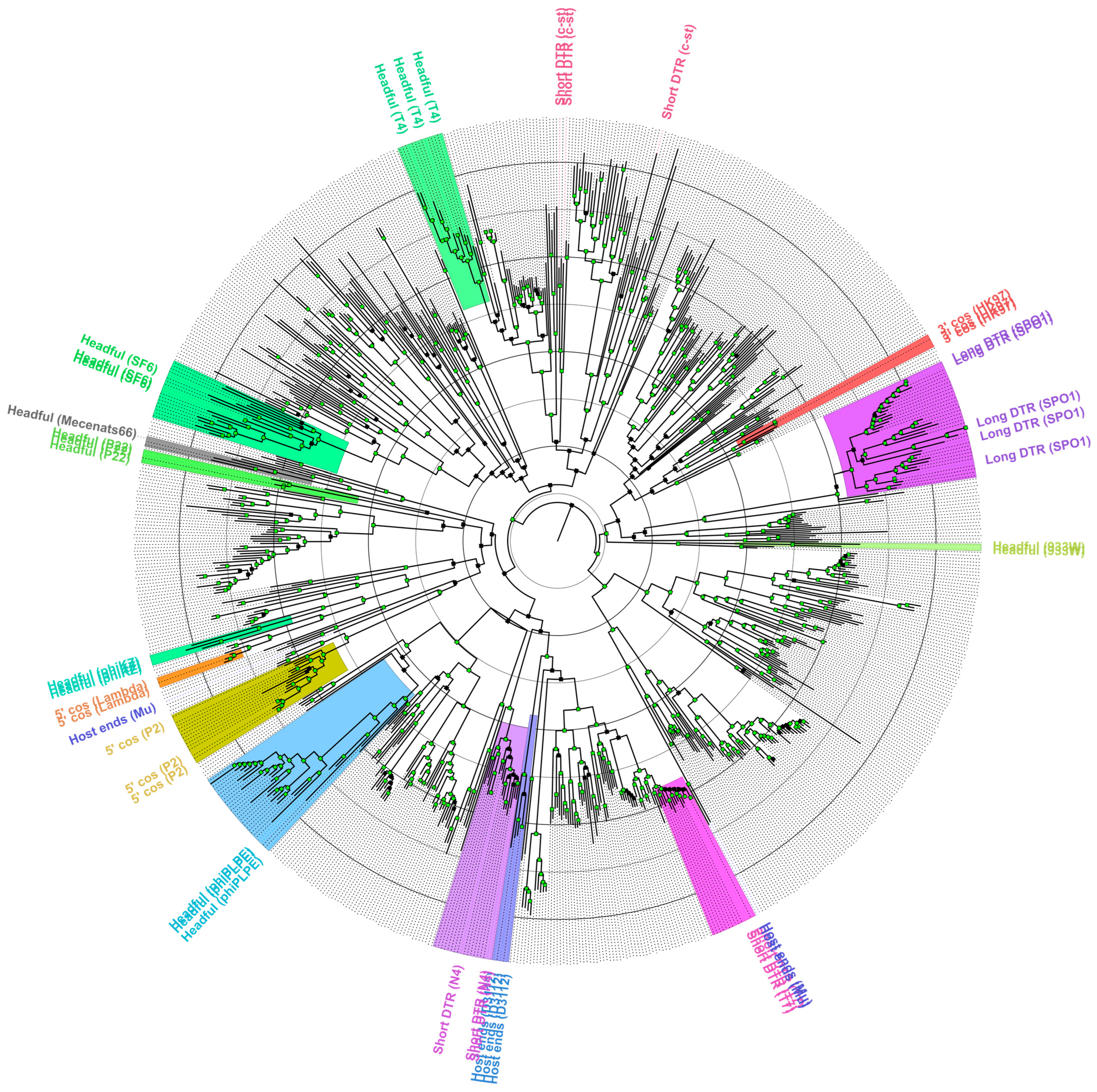
3.4. Robustness of the TerL/Terminase Amino Acid Sequence Phylogeny Reconstructions to Predict the Packaging Strategy of a Novel Phage
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, H.; Zhu, J.; Hu, Q.; Rao, X. Morganella Morganii, a Non-Negligent Opportunistic Pathogen. Int. J. Infect. Dis. 2016, 50, 10–17. [Google Scholar] [CrossRef]
- Safaei, S.; Fatahi-Bafghi, M.; Pouresmaeil, O. Role of Tsukamurella Species in Human Infections: First Literature Review. New Microbes New Infect. 2018, 22, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.P.; Pembroke, J.T. Brevundimonas spp: Emerging Global Opportunistic Pathogens. Virulence 2018, 9, 480–493. [Google Scholar] [CrossRef]
- Lee, J.; Hwang, J.H.; Jo, D.S.; Lee, H.S.; Hwang, J.H. Kluyvera ascorbata as a Pathogen in Adults and Children: Clinical Features and Antibiotic Susceptibilities in a Single Center Study. Jpn. J. Infect. Dis. 2019, 72, 142–148. [Google Scholar] [CrossRef]
- Vouga, M.; Greub, G. Emerging Bacterial Pathogens: The Past and Beyond. Clin. Microbiol. Infect. 2016, 22, 12–21. [Google Scholar] [CrossRef]
- Miller-Ensminger, T.; Garretto, A.; Brenner, J.; Thomas-White, K.; Zambom, A.; Wolfe, A.J.; Putonti, C. Bacteriophages of the Urinary Microbiome. J. Bacteriol. 2018, 200, e00738-17. [Google Scholar] [CrossRef] [PubMed]
- Szafrański, S.P.; Slots, J.; Stiesch, M. The Human Oral Phageome. Periodontol. 2000 2021, 86, 79–96. [Google Scholar] [CrossRef] [PubMed]
- El Karkouri, K.; Ghigo, E.; Raoult, D.; Fournier, P.E. Genomic Evolution and Adaptation of Arthropod-Associated Rickettsia. Sci. Rep. 2022, 12, 3807. [Google Scholar] [CrossRef]
- Emborg, J.; Dalgaard, P.; Ahrens, P. Morganella psychrotolerans sp. nov., a Histamine- Producing Bacterium Isolated from Various Seafoods. Int. J. Syst. Evol. Microbiol. 2006, 56, 2473–2479. [Google Scholar] [CrossRef] [PubMed]
- Emborg, J.; Dalgaard, P. Formation of Histamine and Biogenic Amines in Cold-Smoked Tuna: An Investigation of Psychrotolerant Bacteria from Samples Implicated in Cases of Histamine Fish Poisoning. J. Food Prot. 2006, 69, 897–906. [Google Scholar] [CrossRef]
- Zhu, J.; Rao, X.; Tan, Y.; Xiong, K.; Hu, Z.; Chen, Z.; Jin, X.; Li, S.; Chen, Y.; Hu, F. Identification of Lytic Bacteriophage MmP1, assigned to a New Member of T7-like Phages Infecting Morganella morganii. Genomics 2010, 96, 167–172. [Google Scholar] [CrossRef]
- Oliveira, H.; Pinto, G.; Oliveira, A.; Noben, J.P.; Hendrix, H.; Lavigne, R.; Łobocka, M.; Kropinski, A.M.; Azeredo, J. Characterization and Genomic Analyses of Two Newly Isolated Morganella Phages Define Distant Members among Tevenvirinae and Autographivirinae Subfamilies. Sci. Rep. 2017, 7, 46157. [Google Scholar] [CrossRef]
- Evans, S.; Cobbley, H.; Davis, K.; Divis, T.; Eberhard, N.; Flor, S.; Harris, E.B.; Ben Gordon, J.; Hyer, M.; Larson, W.; et al. Complete Genome Sequences of Five Bacteriophages That Infect Enterobacteriales Hosts. Microbiol. Resour. Announc. 2022, 11, e01223-21. [Google Scholar] [CrossRef]
- Yamaki, S.; Omachi, T.; Kawai, Y.; Yamazaki, K. Characterization of a Novel Morganella morganii Bacteriophage FSP1 Isolated from River Water. FEMS Microbiol. Lett. 2014, 359, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.B.; Feiss, M. The Bacteriophage DNA Packaging Motor. Annu. Rev. Genet. 2008, 42, 647–681. [Google Scholar] [CrossRef]
- Casjens, S.R. The DNA-Packaging Nanomotor of Tailed Bacteriophages. Nat. Rev. Microbiol. 2011, 9, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Dedeo, C.L.; Cingolani, G.; Teschke, C.M. Portal Protein: The Orchestrator of Capsid Assembly for the DsDNA Tailed Bacteriophages and Herpesviruses. Annu. Rev. Virol. 2019, 6, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Aksyuk, A.A.; Rossmann, M.G. Bacteriophage Assembly. Viruses 2011, 3, 172–203. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, H.; Morita, M. Phage DNA Packaging. Genes Cells 2003, 2, 537–545. [Google Scholar] [CrossRef]
- Pruss, G.J.; Calendar, R. Maturation of Bacteriophage P2 DNA. Virology 1978, 86, 454–467. [Google Scholar] [CrossRef]
- George, M.; Bukhari, A.I. Heterogeneous Host DNA Attached to the Left End of Mature Bacteriophage Mu DNA. Nature 1981, 292, 175–176. [Google Scholar] [CrossRef]
- Vahanian, N.; Oh, C.S.; Sippy, J.; Feiss, M. Natural History of a Viral Cohesive End Site: CosN of the λ-like Phages. Virology 2017, 509, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Juhala, R.J.; Ford, M.E.; Duda, R.L.; Youlton, A.; Hatfull, G.F.; Hendrix, R.W. Genomic Sequences of Bacteriophages HK97 and HK022: Pervasive Genetic Mosaicism in the Lambdoid Bacteriophages. J. Mol. Biol. 2000, 299, 27–51. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.B.; Nardone, C.; Hinkle, D.C. Bacteriophage T7 DNA Packaging. III. A “Hairpin” End Formed on T7 Concatemers May Be an Intermediate in the Processing Reaction. J. Mol. Biol. 1990, 216, 939–948. [Google Scholar] [CrossRef]
- Stewart, C.R.; Casjens, S.R.; Cresawn, S.G.; Houtz, J.M.; Smith, A.L.; Ford, M.E.; Peebles, C.L.; Hatfull, G.F.; Hendrix, R.W.; Huang, W.M.; et al. The Genome of Bacillus subtilis Bacteriophage SPO1. J. Mol. Biol. 2009, 388, 48–70. [Google Scholar] [CrossRef] [PubMed]
- Streisinger, G.; Emrich, J.; Stahl, M.M. Chromosome Structure in Phage T4, III. Terminal Redundancy and Length Determination. Proc. Natl. Acad. Sci. USA 1967, 57, 292–295. [Google Scholar] [CrossRef]
- Casjens, S.R.; Gilcrease, E.B. Determining DNA Packaging Strategy by Analysis of the Termini of the Chromosomes in Tailed-Bacteriophage Virions. In Bacteriophages: Methods and Protocols, Molecular and Applied Aspects; Clokie, M.R., Kropinski, A.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; Volume 2, pp. 91–111. [Google Scholar]
- Tye, B.-K.; Huberman, J.A.; Botstein, D. Non-Random Circular Permutation of Phage P22 DNA. J. Mol. Biol. 1974, 85, 501–527. [Google Scholar] [CrossRef]
- Kalinski, A.; Black, L.W. End Structure and Mechanism of Packaging of Bacteriophage T4 DNA. J. Virol. 1986, 58, 951–954. [Google Scholar] [CrossRef]
- Bjornsti, M.A.; Reilly, B.E.; Anderson, D.L. Morphogenesis of Bacteriophage Phi 29 of Bacillus Subtilis: Oriented and Quantized in Vitro Packaging of DNA Protein Gp3. J. Virol. 1983, 45, 383–396. [Google Scholar] [CrossRef]
- Garneau, J.R.; Depardieu, F.; Fortier, L.C.; Bikard, D.; Monot, M. PhageTerm: A Tool for Fast and Accurate Determination of Phage Termini and Packaging Mechanism Using Next-Generation Sequencing Data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef]
- Li, S.; Fan, H.; An, X.; Fan, H.; Jiang, H.; Chen, Y.; Tong, Y. Scrutinizing Virus Genome Termini by High-Throughput Sequencing. PLoS ONE 2014, 9, e85806. [Google Scholar] [CrossRef] [PubMed]
- Casjens, S.R.; Gilcrease, E.B.; Winn-Stapley, D.A.; Schicklmaier, P.; Schmieger, H.; Pedulla, M.L.; Ford, M.E.; Houtz, J.M.; Hatfull, G.F.; Hendrix, R.W. The Generalized Transducing Salmonella Bacteriophage ES18: Complete Genome Sequence and DNA Packaging Strategy. J. Bacteriol. 2005, 187, 1091–1104. [Google Scholar] [CrossRef] [PubMed]
- Merrill, B.D.; Ward, A.T.; Grose, J.H.; Hope, S. Software-Based Analysis of Bacteriophage Genomes, Physical Ends, and Packaging Strategies. BMC Genom. 2016, 17, 679. [Google Scholar] [CrossRef]
- Esterman, E.S.; I Wolf, Y.; Kogay, R.; Koonin, E.V.; Zhaxybayeva, O. Evolution of DNA Packaging in Gene Transfer Agents. Virus Evol. 2021, 7, veab015. [Google Scholar] [CrossRef] [PubMed]
- Zrelovs, N.; Cernooka, E.; Dislers, A.; Kazaks, A. Isolation and Characterization of the Novel Virgibacillus-Infecting Bacteriophage Mimir87. Arch. Virol. 2020, 165, 737–741. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC—A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 17 November 2021).
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner. In Proceedings of the Conference 9th Annual Genomics of Energy & Environment Meeting, Walnut Creek, CA, USA, 17–20 March 2014. [Google Scholar]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving Bacterial Genome Assemblies from Short and Long Sequencing Reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying Bacterial Genes and Endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Besemer, J.; Borodovsky, M. GeneMark: Web Software for Gene Finding in Prokaryotes, Eukaryotes and Viruses. Nucleic Acids Res. 2005, 33, 451–454. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARAGORN, a Program to Detect TRNA Genes and TmRNA Genes in Nucleotide Sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. TRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Bryant, S.H. CD-Search: Protein Domain Annotations on the Fly. Nucleic Acids Res. 2004, 32, W327–W331. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred Interactive Server for Protein Homology Detection and Structure Prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef]
- Amin, M.R.; Yurovsky, A.; Chen, Y.; Skiena, S.; Futcher, B. Re-Annotation of 12,495 Prokaryotic 16s Rrna 3’ Ends and Analysis of Shine-Dalgarno and Anti-Shine-Dalgarno Sequences. PLoS ONE 2018, 13, e0202767. [Google Scholar] [CrossRef]
- Starmer, J.; Stomp, A.; Vouk, M.; Bitzer, D. Predicting Shine-Dalgarno Sequence Locations Exposes Genome Annotation Errors. PLoS Comput. Biol. 2006, 2, 454–466. [Google Scholar] [CrossRef]
- Weisburg, W.G.; Barns, S.M.; Pelletier, D.A.; Lane, D.J. 16S Ribosomal DNA Amplification for Phylogenetic Study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA Sequencing with Chain-Terminating Inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A Taxonomically United Database of 16S RRNA Gene Sequences and Whole-Genome Assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and Tools for High Throughput RRNA Analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Lu, S.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; et al. CDD: A Conserved Domain Database for the Functional Annotation of Proteins. Nucleic Acids Res. 2011, 39, 225–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities Of prokaryote-infecting viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Gilchrist, C.L.M.; Chooi, Y.H. Clinker & Clustermap.Js: Automatic Generation of Gene Cluster Comparison Figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, L.; Sun, E.; Song, J.; Wu, B. Characterisation of a Newly Detected Bacteriophage Infecting Bordetella Bronchiseptica in Swine. Arch. Virol. 2019, 164, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A Genome Comparison Visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Accession | Phage | Genome Length (kbp) | GC% Content | CDS Number | Family | Genus | Isolation Source | Host | Reference |
|---|---|---|---|---|---|---|---|---|---|
| EU652770 | Morganella phage MmP1 | 38.457 | 46.52 | 49 | Autographiviridae | Minipunavirus | Sewage | Morganella morganii | [11] |
| KX078568 | Morganella phage vB_MmoP_MP2 | 39.616 | 46.91 | 55 | Autographiviridae | Minipunavirus | Sewage | Morganella sp. | [12] |
| KX078569 | Morganella phage vB_MmoM_MP1 | 163.201 | 34.75 | 271 | Straboviridae | Gualtarvirus | Sewage | Morganella sp. | [12] |
| KY653118 | Morganella phage IME1369_01 | 47.739 | 45.82 | 67 | Siphoviridae * | not defined | Host (prophage) | Morganella morganii IME1369 | N/A |
| KY653119 | Morganella phage IME1369_02 | 39.387 | 50.02 | 60 | Siphoviridae * | not defined | Host (prophage) | Morganella morganii IME1369 | N/A |
| OK499982 | Morganella phage vB_MmoP_Lilpapawes | 39.168 | 47.06 | 57 | Autographiviridae | Minipunavirus | Sewage | Morganella morganii | [13] |
| OK499989 | Morganella phage vB_MmoM_Rgz1 | 165.808 | 34.71 | 275 | Straboviridae | Gualtarvirus | Sewage | Morganella morganii | [13] |
| OL614104 | Morganella phage Mecenats66 | 86.193 | 49.07 | 123 | Myoviridae * | not defined | Insect gut | Morganella sp. | This study |
| N/A | Morganella phage FSP1 | ~45.6–49.4 | N/A | N/A | Myoviridae * | N/A | River water | Morganella morganii ssp. morganii | [14] |
| Primer | Primer Sequence (5’–3’) | Coordinates in Mecenats66 Genome (Bases) |
|---|---|---|
| ph66_C_Fw | GCAGCAGGATCGTTAAGTCC | 85,939–85,958 |
| ph66_N_Rv | CTAGATCGCATCGATTGCAG | 334–353 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zrelovs, N.; Jansons, J.; Dislers, A.; Kazaks, A. Morganella Phage Mecenats66 Utilizes an Evolutionarily Distinct Subtype of Headful Genome Packaging with a Preferred Packaging Initiation Site. Microorganisms 2022, 10, 1799. https://doi.org/10.3390/microorganisms10091799
Zrelovs N, Jansons J, Dislers A, Kazaks A. Morganella Phage Mecenats66 Utilizes an Evolutionarily Distinct Subtype of Headful Genome Packaging with a Preferred Packaging Initiation Site. Microorganisms. 2022; 10(9):1799. https://doi.org/10.3390/microorganisms10091799
Chicago/Turabian StyleZrelovs, Nikita, Juris Jansons, Andris Dislers, and Andris Kazaks. 2022. "Morganella Phage Mecenats66 Utilizes an Evolutionarily Distinct Subtype of Headful Genome Packaging with a Preferred Packaging Initiation Site" Microorganisms 10, no. 9: 1799. https://doi.org/10.3390/microorganisms10091799
APA StyleZrelovs, N., Jansons, J., Dislers, A., & Kazaks, A. (2022). Morganella Phage Mecenats66 Utilizes an Evolutionarily Distinct Subtype of Headful Genome Packaging with a Preferred Packaging Initiation Site. Microorganisms, 10(9), 1799. https://doi.org/10.3390/microorganisms10091799