
Advanced Glycation End Products in Health and Disease



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
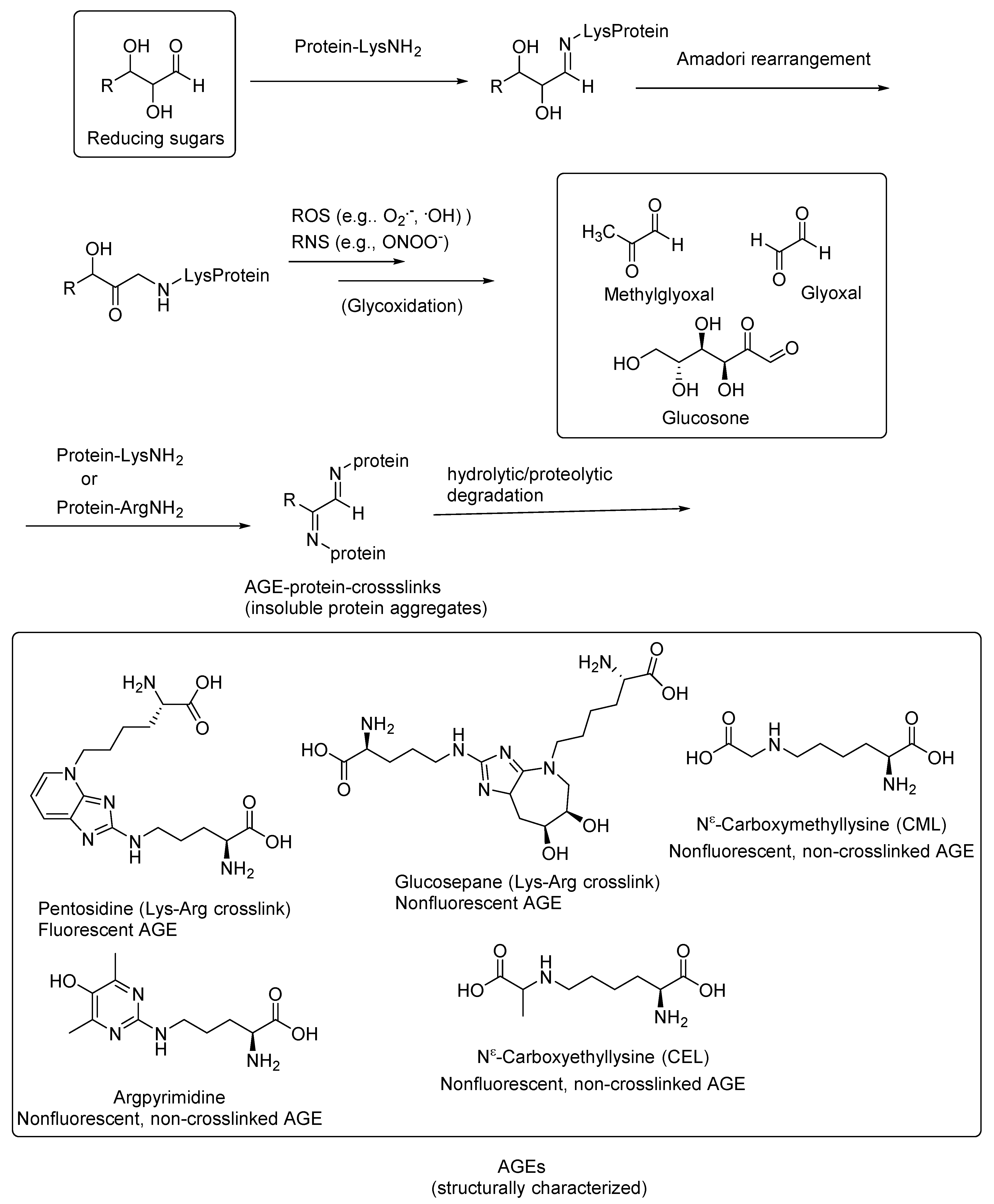
:1. Introduction
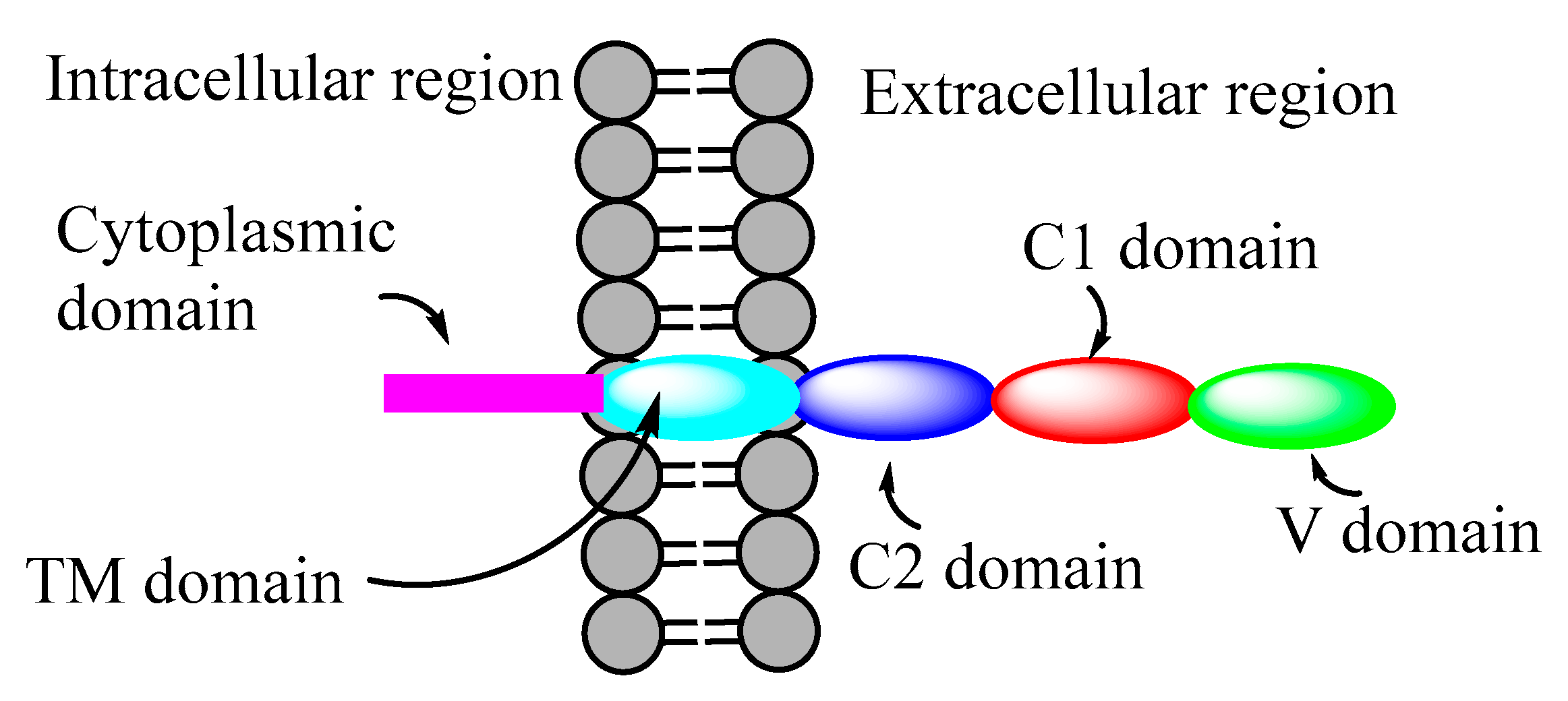
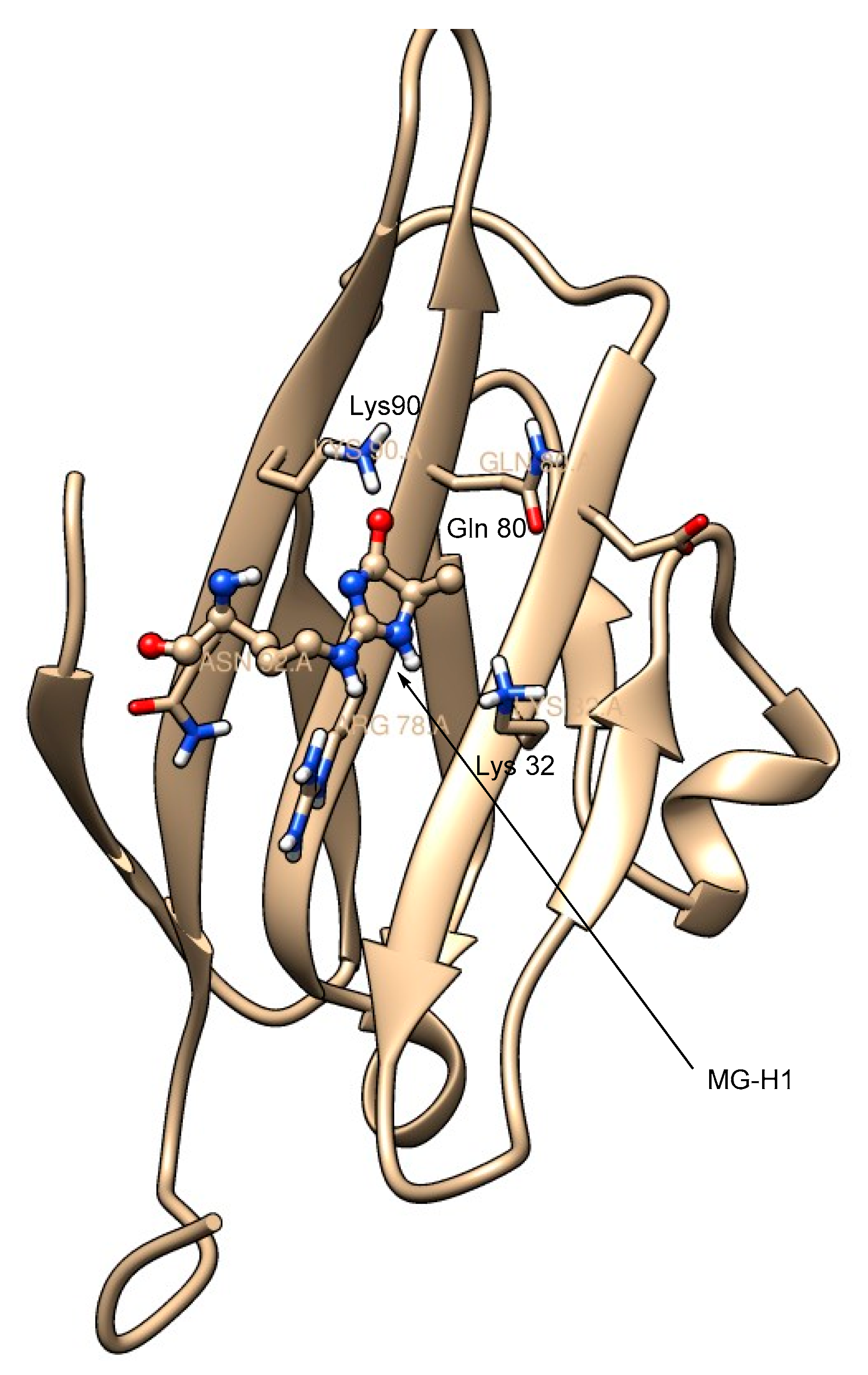
2. Receptors for Advanced Glycation End Products (RAGE)
3. AGE Inhibitors
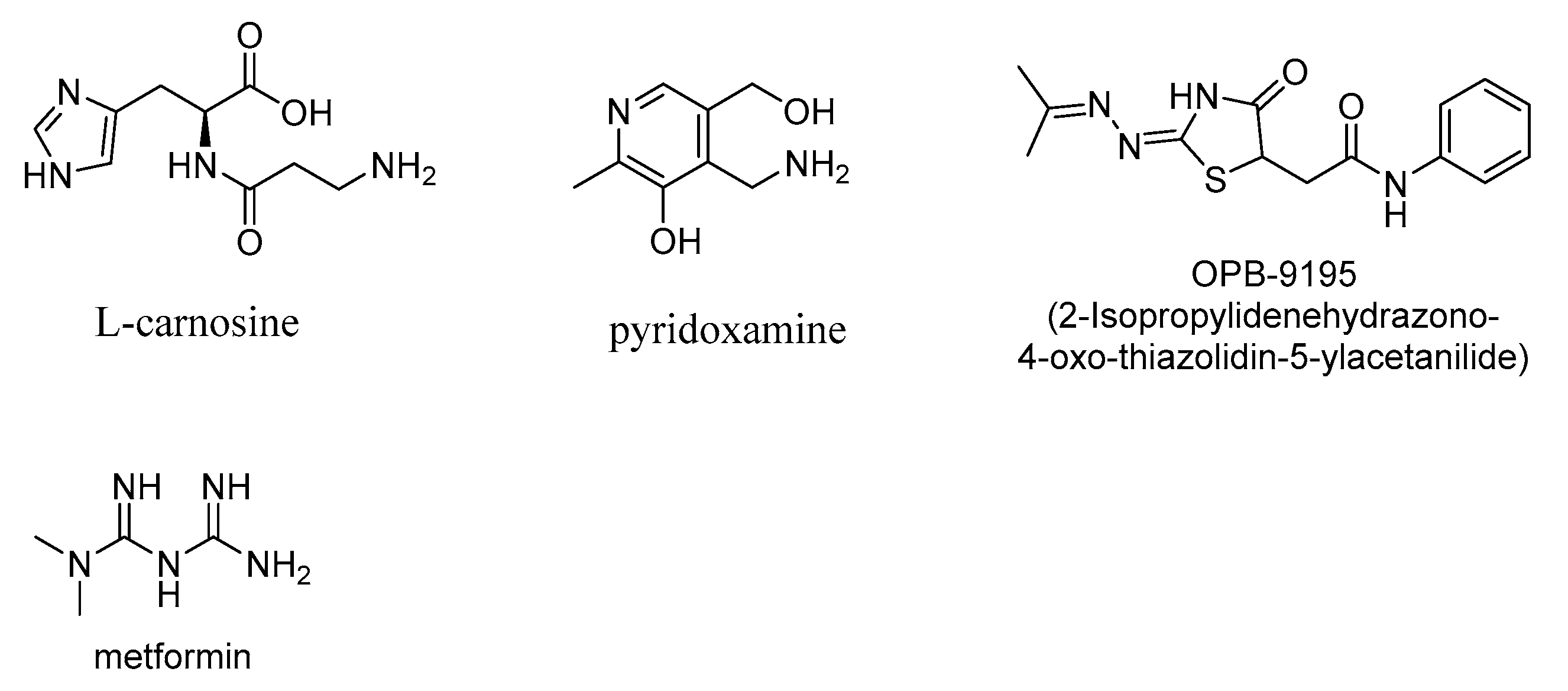
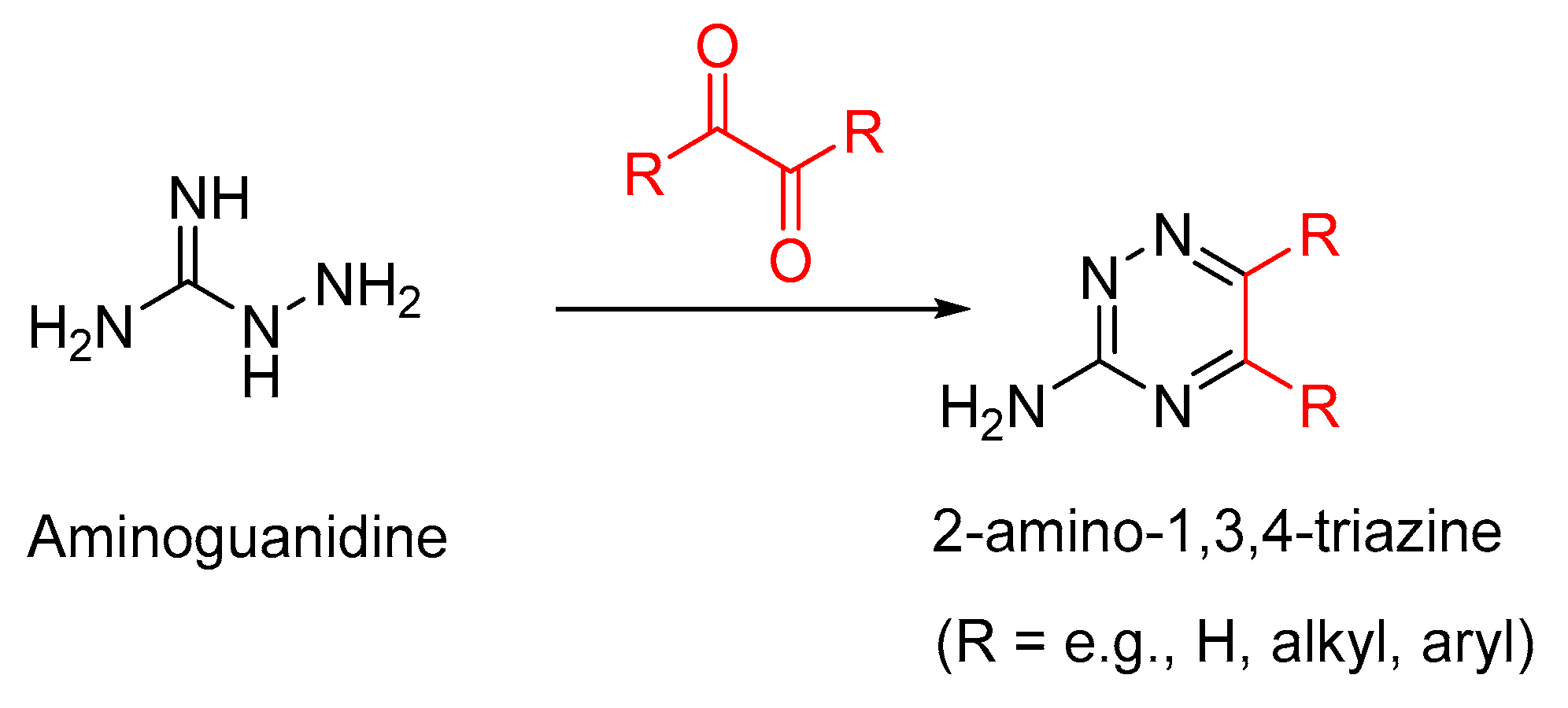
3.1. Aminoguanidine
3.2. Pyridoxamine
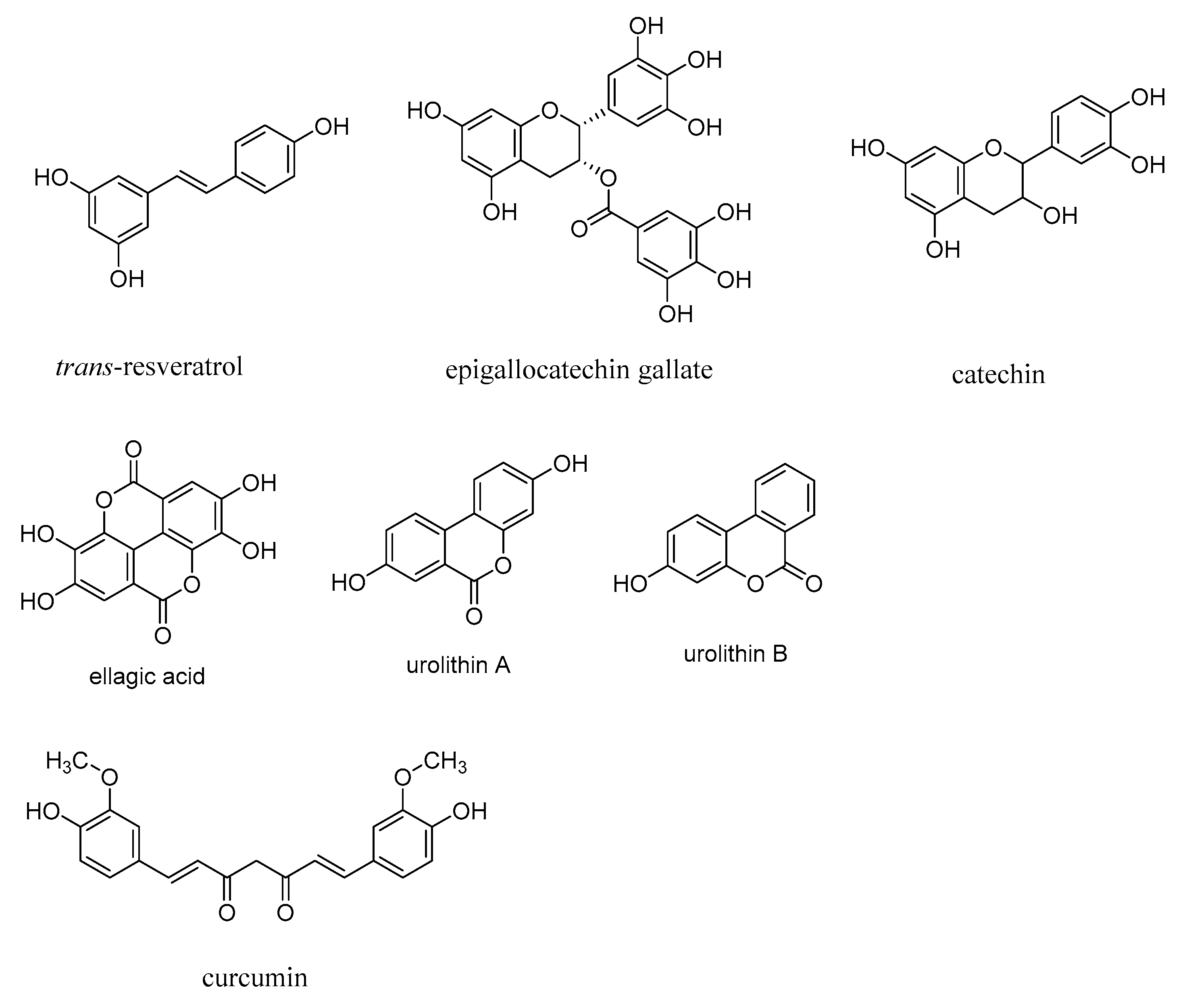
3.3. Polyphenols as AGE Inhibitors
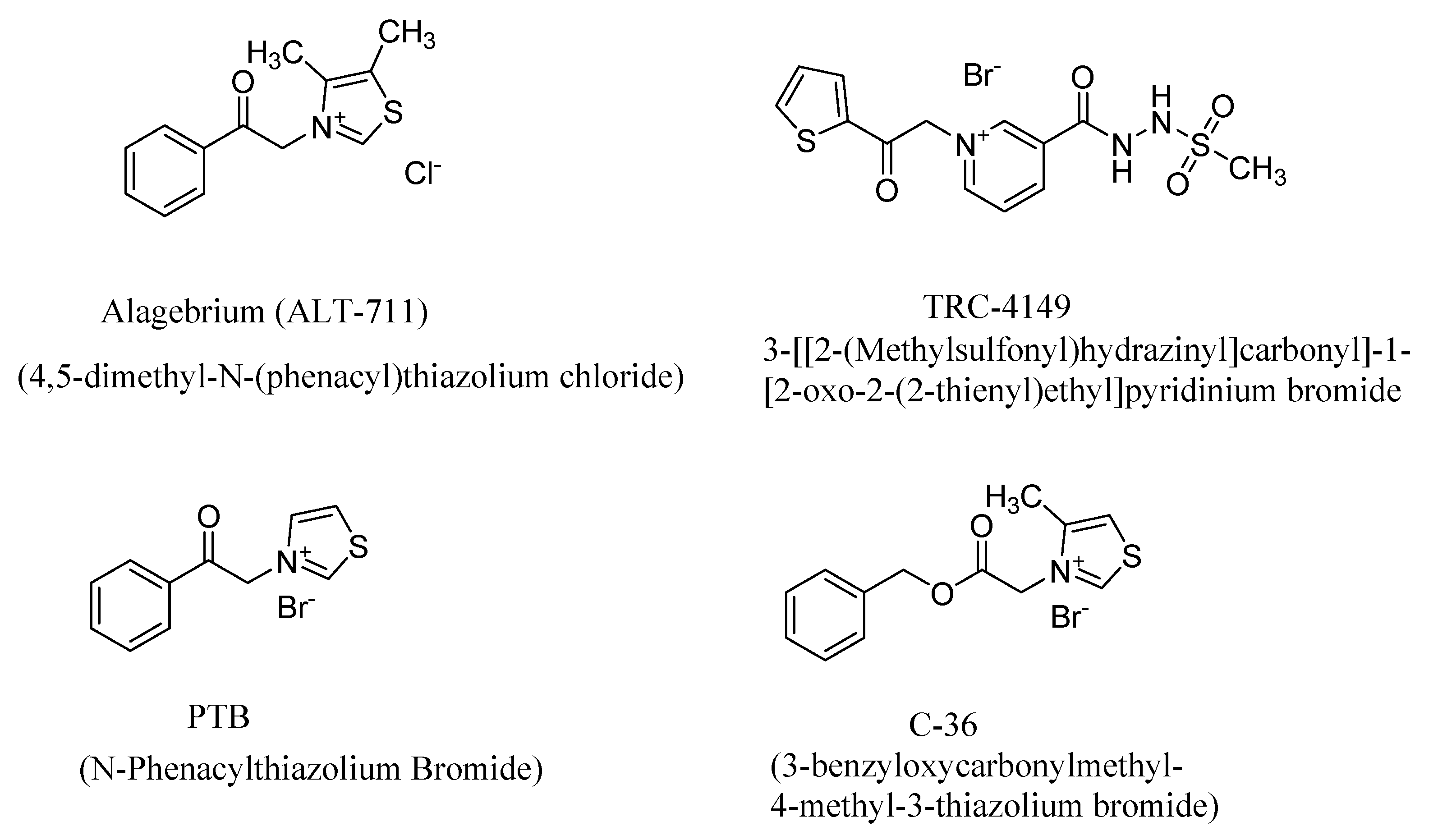
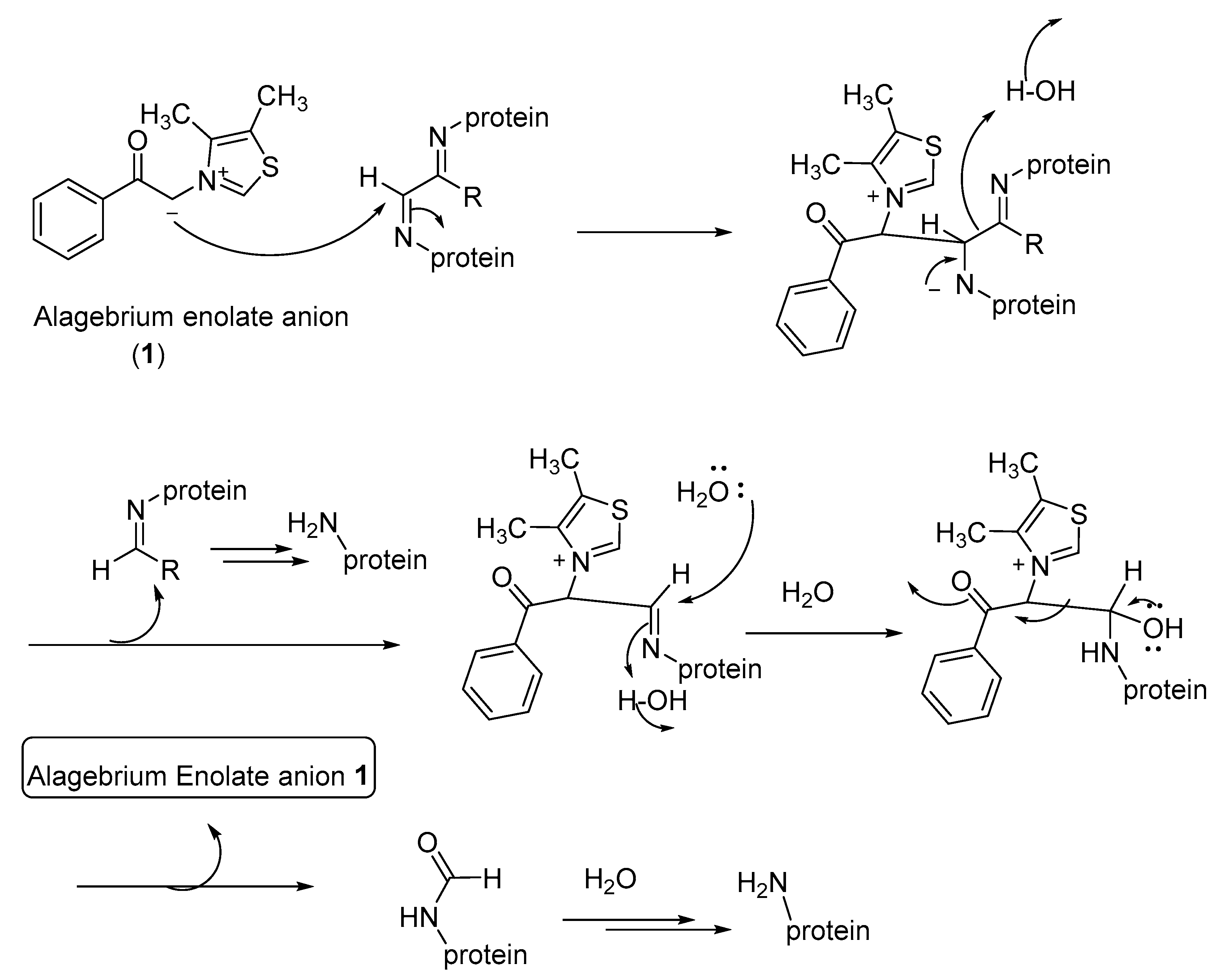
4. AGE Breakers
5. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Reddy, V.P.; Beyaz, A. Inhibitors of the Maillard reaction and AGE breakers as therapeutics for multiple diseases. Drug Discov. Today 2006, 11, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. 2010, 345, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.P.; Beyaz, A.; Perry, G.; Cooke, M.S.; Sayre, L.M.; Smith, M.A. The role of oxidative damage to nucleic acids in the pathogenesis of neurological diseases. In Oxidative Damage to Nucleic Acids; Evans, M.D., Cooke, M.S., Eds.; Landes Bioscience and Springer Science+Business Media, LLC: Austin, TX, USA, 2007; pp. 123–140. [Google Scholar]
- Ott, C.; Jacobs, K.; Haucke, E.; Santos, A.N.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.K.; Bierhaus, A.; Schiekofer, S.; Tritschler, H.; Ziegler, R.; Nawroth, P.P. The role of oxidative stress and NF-κB activation in late diabetic complications. BioFactors 1999, 10, 157–167. [Google Scholar] [CrossRef]
- Menini, S.; Iacobini, C.; Vitale, M.; Pesce, C.; Pugliese, G. Diabetes and pancreatic cancer-a dangerous liaison relying on carbonyl stress. Cancers 2021, 13, 313. [Google Scholar] [CrossRef]
- Koschinsky, T.; He, C.-J.; Mitsuhashi, T.; Bucala, R.; Liu, C.; Buenting, C.; Heitmann, K.; Vlassara, H. Orally absorbed reactive glycation products (glycotoxins): An environmental risk factor in diabetic nephropathy. Proc. Natl. Acad. Sci. USA. 1997, 94, 6474–6479. [Google Scholar] [CrossRef]
- Fotheringham, A.K.; Gallo, L.A.; Borg, D.J.; Forbes, J.M. Advanced Glycation End Products (AGEs) and Chronic Kidney Disease: Does the Modern Diet AGE the Kidney? Nutrients 2022, 14, 2675. [Google Scholar] [CrossRef]
- Forbes, J.M.; Coughlan, M.T.; Cooper, M.E. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 2008, 57, 1446–1454. [Google Scholar] [CrossRef]
- Ahmed, N. Advanced glycation endproducts-role in pathology of diabetic complications. Diabetes Res. Clin. Pract. 2005, 67, 3–21. [Google Scholar] [CrossRef]
- Stratmann, B. Dicarbonyl Stress in Diabetic Vascular Disease. Int. J. Mol. Sci. 2022, 23, 6186. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Hori, O.; Brett, J.; Yan, S.D.; Wautier, J.-L.; Stern, D. Cellular receptors for advanced glycation end products: Implications for induction of oxidant stress and cellular dysfunction in the pathogenesis of vascular lesions. Arter. Thromb. 1994, 14, 1521. [Google Scholar] [CrossRef] [PubMed]
- Pinto, R.S.; Ferreira, G.S.; Silvestre, G.C.R.; Santana, M.d.F.M.; Nunes, V.S.; Ledesma, L.; Pinto, P.R.; de Assis, S.I.S.; Machado, U.F.; da Silva, E.S.; et al. Plasma advanced glycation end products and soluble receptor for advanced glycation end products as indicators of sterol content in human carotid atherosclerotic plaques. Diabetes Vasc. Dis. Res. 2022, 19, 14791641221085269. [Google Scholar] [CrossRef] [PubMed]
- Kislinger, T.; Fu, C.; Huber, B.; Qu, W.; Taguchi, A.; Yan, S.D.; Hofmann, M.; Yan, S.F.; Pischetsrieder, M.; Stern, D.; et al. Nε-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J. Biol. Chem. 1999, 274, 31740–31749. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Castellani, R.J.; Harris, P.L.R.; Sayre, L.M.; Fujii, J.; Taniguchi, N.; Vitek, M.P.; Founds, H.; Atwood, C.S.; Perry, G.; Smith, M.A. Active glycation in neurofibrillary pathology of Alzheimer’s disease: Nε-(Carboxymethyl) lysine and hexitol-lysine. Free. Radic. Biol. Med. 2001, 31, 175–180. [Google Scholar] [CrossRef]
- Horie, K.; Miyata, T.; Yasuda, T.; Takeda, A.; Yasuda, Y.; Maeda, K.; Sobue, G.; Kurokawa, K. Immunohistochemical localization of advanced glycation end products, pentosidine, and carboxymethyllysine in lipofuscin pigments of Alzheimer’s disease and aged neurons. Biochem. Biophys. Res. Commun. 1997, 236, 327–332. [Google Scholar] [CrossRef]
- Haddad, M.; Perrotte, M.; Landri, S.; Lepage, A.; Fülöp, T.; Ramassamy, C. Circulating and Extracellular Vesicles Levels of N-(1-Carboxymethyl)-L-Lysine (CML) Differentiate Early to Moderate Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 69, 751–762. [Google Scholar] [CrossRef]
- Dei, R.; Takeda, A.; Niwa, H.; Li, M.; Nakagomi, Y.; Watanabe, M.; Inagaki, T.; Washimi, Y.; Yasuda, Y.; Horie, K.; et al. Lipid peroxidation and advanced glycation end products in the brain in normal aging and in Alzheimer’s disease. Acta Neuropathol. 2002, 104, 113–122. [Google Scholar] [CrossRef]
- Kikuchi, S.; Shinpo, K.; Ogata, A.; Tsuji, S.; Takeuchi, M.; Makita, Z.; Tashiro, K. Detection of Nε-(carboxymethyl)lysine (CML) and non-CML advanced glycation end-products in the anterior horn of amyotrophic lateral sclerosis spinal cord. Amyotrophic Lateral Scler. Other Mot. Neuron Disord. 2002, 3, 63–68. [Google Scholar]
- Juranek, J.K.; Daffu, G.K.; Wojtkiewicz, J.; Lacomis, D.; Kofler, J.; Schmidt, A.M. Receptor for advanced glycation end products and its inflammatory ligands are upregulated in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2015, 9, 485/1–485/12. [Google Scholar] [CrossRef]
- Chou, S.M.; Wang, H.S.; Taniguchi, A.; Bucala, R. Advanced glycation endproducts in neurofilament conglomeration of motoneurons in familial and sporadic amyotrophic lateral sclerosis. Mol. Med. 1998, 4, 324–332. [Google Scholar] [CrossRef] [PubMed]
- MacLean, M.; Juranek, J.; Cuddapah, S.; Lopez-Diez, R.; Ruiz, H.H.; Hu, J.; Frye, L.; Li, H.; Gugger, P.F.; Schmidt, A.M. Microglia RAGE exacerbates the progression of neurodegeneration within the SOD1G93A murine model of amyotrophic lateral sclerosis in a sex-dependent manner. J. Neuroinflamm. 2021, 18, 139. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.A.; Sonnet, P. A novel multi-target strategy to attenuate the progression of Parkinson’s disease by diamine hybrid AGE/ALE inhibitor. Future Med. Chem. 2021, 13, 2185–2200. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.; Vicente Miranda, H.; Outeiro, T.F. Alpha-Synuclein Glycation and the Action of Anti-Diabetic Agents in Parkinson’s Disease. J. Parkinson’s Dis. 2018, 8, 33–43. [Google Scholar] [CrossRef]
- Chrysanthou, M.; Estruch, I.M.; Rietjens, I.M.C.M.; Wichers, H.J.; Hoppenbrouwers, T. In Vitro Methodologies to Study the Role of Advanced Glycation End Products (AGEs) in Neurodegeneration. Nutrients 2022, 14, 363. [Google Scholar] [CrossRef]
- Saglam, E.; Zirh, S.; Aktas, C.C.; Muftuoglu, S.F.; Bilginer, B. Papaverine provides neuroprotection by suppressing neuroinflammation and apoptosis in the traumatic brain injury via RAGE- NF-<kappa>B pathway. J. Neuroimmunol. 2021, 352, 577476. [Google Scholar] [CrossRef]
- Gao, T.-L.; Yuan, X.-T.; Yang, D.; Dai, H.-L.; Wang, W.-J.; Peng, X.; Shao, H.-J.; Jin, Z.-F.; Fu, Z.-J. Expression of HMGB1 and RAGE in rat and human brains after traumatic brain injury. J. Trauma Acute Care Surg. 2012, 72, 643–649. [Google Scholar] [CrossRef]
- Yanagisawa, K.; Makita, Z.; Shiroshita, K.; Ueda, T.; Fusegawa, T.; Kuwajima, S.; Takeuchi, M.; Koike, T. Specific fluorescence assay for advanced glycation end products in blood and urine of diabetic patients. Metab. Clin. Exp. 1998, 47, 1348–1353. [Google Scholar] [CrossRef]
- Nagai, R.; Shirakawa, J.-I.; Ohno, R.-I.; Hatano, K.; Sugawa, H.; Arakawa, S.; Ichimaru, K.; Kinoshita, S.; Sakata, N.; Nagai, M. Antibody-based detection of advanced glycation end-products: Promises vs. limitations. Glycoconj. J. 2016, 33, 545–552. [Google Scholar] [CrossRef]
- Reddy, V.P.; Obrenovich, M.E.; Atwood, C.S.; Perry, G.; Smith, M.A. Involvement of Maillard reactions in Alzheimer disease. Neurotox. Res. 2002, 4, 191–209. [Google Scholar] [CrossRef]
- Boteva, E.; Mironova, R. Maillard reaction and aging: Can bacteria shed light on the link? Biotechnol. Biotechnol. Equip. 2019, 33, 481–497. [Google Scholar] [CrossRef]
- Markesbery, W.R. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar] [CrossRef]
- Vitek, M.P.; Bhattacharya, K.; Glendening, J.M.; Stopa, E.; Vlassara, H.; Bucala, R.; Manogue, K.; Cerami, A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 4766. [Google Scholar] [CrossRef]
- Wang, J.; Cai, W.; Yu, J.; Liu, H.; He, S.; Zhu, L.; Xu, J. Dietary Advanced Glycation End Products Shift the Gut Microbiota Composition and Induce Insulin Resistance in Mice. Diabetes Metab. Syndr. Obes. Targets Ther. 2022, 15, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Kazi, R.S.; Banarjee, R.M.; Deshmukh, A.B.; Patil, G.V.; Jagadeeshaprasad, M.G.; Kulkarni, M.J. Glycation inhibitors extend yeast chronological lifespan by reducing advanced glycation end products and by back regulation of proteins involved in mitochondrial respiration. J. Proteom. 2017, 156, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, S.F.; Seo, Y.; Allen, C.; Ghanta, K.S.; Finkel, S.; Tissenbaum, H.A. Bacterial processing of glucose modulates C. elegans lifespan and healthspan. Sci. Rep. 2021, 11, 5931. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, M.; Auerbach, C.; Mueller, N.; Samuel, P.; Kammann, S.; Beer, F.; Gunzer, F.; Henle, T. Metabolization of the Advanced Glycation End Product N-ε-Carboxymethyllysine (CML) by Different Probiotic E. coli Strains. J. Agric. Food Chem. 2019, 67, 1963–1972. [Google Scholar] [CrossRef]
- Kim, N.Y.; Goddard, T.N.; Sohn, S.; Spiegel, D.A.; Crawford, J.M. Biocatalytic Reversal of Advanced Glycation End Product Modification. ChemBioChem 2019, 20, 2402–2410. [Google Scholar] [CrossRef]
- Mastrocola, R.; Collotta, D.; Gaudioso, G.; Le Berre, M.; Cento, A.S.; Alves, G.F.; Chiazza, F.; Verta, R.; Bertocchi, I.; Manig, F.; et al. Effects of exogenous dietary advanced glycation end products on the cross-talk mechanisms linking microbiota to metabolic inflammation. Nutrients 2020, 12, 2497. [Google Scholar] [CrossRef]
- Smith, A.J.; Advani, J.; Brock, D.C.; Nellissery, J.; Gumerson, J.; Dong, L.; Aravind, L.; Kennedy, B.; Swaroop, A. GATD3A, a mitochondrial deglycase with evolutionary origins from gammaproteobacteria, restricts the formation of advanced glycation end products. BMC Biol. 2022, 20, 68. [Google Scholar] [CrossRef]
- Bongarzone, S.; Savickas, V.; Luzi, F.; Gee, A.D. Targeting the Receptor for Advanced Glycation Endproducts (RAGE): A Medicinal Chemistry Perspective. J. Med. Chem. 2017, 60, 7213–7232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.J.; Park, J.H. Receptor for Advanced Glycation Endproducts (RAGE), Its Ligands, and Soluble RAGE: Potential Biomarkers for Diagnosis and Therapeutic Targets for Human Renal Diseases. Genom. Inf. 2013, 11, 224–229. [Google Scholar] [CrossRef]
- Radi, R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. USA 2004, 101, 4003–4008. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Mulrennan, S.; Baltic, S.; Aggarwal, S.; Wood, J.; Miranda, A.; Frost, F.; Kaye, J.; Thompson, P.J. The Role of Receptor for Advanced Glycation End Products in Airway Inflammation in CF and CF related Diabetes. Sci. Rep. 2015, 5, 8931. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Agrawal, D.K. Therapeutic potential of targeting the receptor for advanced glycation end products (RAGE) by sma11molecule inhibitors. Drug Dev. Res. 2022, 1–13. [Google Scholar] [CrossRef]
- Xue, J.; Rai, V.; Singer, D.; Chabierski, S.; Xie, J.; Reverdatto, S.; Burz, D.S.; Schmidt, A.M.; Hoffmann, R.; Shekhtman, A. Advanced Glycation End Product Recognition by the Receptor for AGEs. Structure 2011, 19, 722–732. [Google Scholar] [CrossRef]
- Xue, J.; Ray, R.; Singer, D.; Bohme, D.; Burz, D.S.; Rai, V.; Hoffmann, R.; Shekhtman, A. The Receptor for Advanced Glycation End Products (RAGE) Specifically Recognizes Methylglyoxal-Derived AGEs. Biochemistry 2014, 53, 3327–3335. [Google Scholar] [CrossRef]
- Ostendorp, T.; Weibel, M.; Leclerc, E.; Kleinert, P.; Kroneck, P.M.H.; Heizmann, C.W.; Fritz, G. Expression and purification of the soluble isoform of human receptor for advanced glycation end products (sRAGE) from Pichia pastoris. Biochem. Biophys. Res. Commun. 2006, 347, 4–11. [Google Scholar] [CrossRef]
- Prasad, K. AGE-RAGE stress: A changing landscape in pathology and treatment of Alzheimer’s disease. Mol. Cell. Biochem. 2019, 459, 95–112. [Google Scholar] [CrossRef]
- Juranek, J.; Mukherjee, K.; Kordas, B.; Załęcki, M.; Korytko, A.; Zglejc-Waszak, K.; Szuszkiewicz, J.; Banach, M. Role of RAGE in the Pathogenesis of Neurological Disorders. Neurosci. Bull. 2022. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Ma, Y.; Zeng, Z.; Yin, Q.; Hong, Y.; Hou, X.; Liu, X. RAGE-Specific Inhibitor FPS-ZM1 Attenuates AGEs-Induced Neuroinflammation and Oxidative Stress in Rat Primary Microglia. Neurochem. Res. 2017, 42, 2902–2911. [Google Scholar] [CrossRef]
- Liu, Y.; Shen, W.; Chen, Q.; Cao, Q.; Di, W.; Lan, R.; Chen, Z.; Bai, J.; Han, Z.; Xu, W. Inhibition of RAGE by FPS-ZM1 alleviates renal injury in spontaneously hypertensive rats. Eur. J. Pharmacol. 2020, 882, 173228. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Shen, C.; Yin, Q.; Sun, M.; Ma, Y.; Liu, X. Effects of RAGE-Specific Inhibitor FPS-ZM1 on Amyloid-β Metabolism and AGEs-Induced Inflammation and Oxidative Stress in Rat Hippocampus. Neurochem. Res. 2016, 41, 1192–1199. [Google Scholar] [CrossRef]
- Burstein, A.H.; Sabbagh, M.; Andrews, R.; Valcarce, C.; Dunn, I.; Altstiel, L. Development of Azeliragon, an Oral Small Molecule Antagonist of the Receptor for Advanced Glycation Endproducts, for the Potential Slowing of Loss of Cognition in Mild Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2018, 5, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.-K.; Chao, S.-P.; Hu, C.-J. Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef]
- Pinkas, A.; Aschner, M. Advanced Glycation End-Products and Their Receptors: Related Pathologies, Recent Therapeutic Strategies, and a Potential Model for Future Neurodegeneration Studies. Chem. Res. Toxicol. 2016, 29, 707–714. [Google Scholar] [CrossRef]
- Tan, K.C.B.; Chow, W.S.; Tso, A.W.K.; Xu, A.; Tse, H.F.; Hoo, R.L.C.; Betteridge, D.J.; Lam, K.S.L. Thiazolidinedione increases serum soluble receptor for advanced glycation end-products in type 2 diabetes. Diabetologia 2007, 50, 1819–1825. [Google Scholar] [CrossRef]
- Sabbatinelli, J.; Castiglione, S.; Macri, F.; Giuliani, A.; Ramini, D.; Vinci, M.C.; Tortato, E.; Bonfigli, A.R.; Olivieri, F.; Raucci, A. Circulating levels of AGEs and soluble RAGE isoforms are associated with all-cause mortality and development of cardiovascular complications in type 2 diabetes: A retrospective cohort study. Cardiovasc. Diabetol. 2022, 21, 95. [Google Scholar] [CrossRef]
- Colhoun, H.M.; Betteridge, D.J.; Durrington, P.; Hitman, G.; Neil, A.; Livingstone, S.; Charlton-Menys, V.; Bao, W.; DeMicco, D.A.; Preston, G.M.; et al. Total soluble and endogenous secretory receptor for advanced glycation end products as predictive biomarkers of coronary heart disease risk in patients with type 2 diabetes: An analysis from the CARDS trial. Diabetes 2011, 60, 2379–2385. [Google Scholar] [CrossRef]
- Fujisawa, K.; Katakami, N.; Kaneto, H.; Naka, T.; Takahara, M.; Sakamoto, F.; Irie, Y.; Miyashita, K.; Kubo, F.; Yasuda, T.; et al. Circulating soluble RAGE as a predictive biomarker of cardiovascular event risk in patients with type 2 diabetes. Atherosclerosis 2013, 227, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Legaard, G.E.; Feineis, C.S.; Johansen, M.Y.; Hansen, K.B.; Vaag, A.A.; Larsen, E.L.; Poulsen, H.E.; Almdal, T.P.; Karstoft, K.; Pedersen, B.K.; et al. Effects of an exercise-based lifestyle intervention on systemic markers of oxidative stress and advanced glycation endproducts in persons with type 2 diabetes: Secondary analysis of a randomised clinical trial. Free Radic. Biol. Med. 2022, 188, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Adeshara, K.A.; Bangar, N.; Diwan, A.G.; Tupe, R.S. Plasma glycation adducts and various RAGE isoforms are intricately associated with oxidative stress and inflammatory markers in type 2 diabetes patients with vascular complications. Diabetes Metab. Syndr. 2022, 16, 102441. [Google Scholar] [CrossRef] [PubMed]
- Steenbeke, M.; De Decker, I.; Marchand, S.; Glorieux, G.; Van Biesen, W.; Lapauw, B.; Delanghe, J.R.; Speeckaert, M.M. Dietary Advanced Glycation End Products in an Elderly Population with Diabetic Nephropathy: An Exploratory Investigation. Nutrients 2022, 14, 1818. [Google Scholar] [CrossRef]
- Pratte, K.A.; Curtis, J.L.; Kechris, K.; Couper, D.; Cho, M.H.; Silverman, E.K.; DeMeo, D.L.; Sciurba, F.C.; Zhang, Y.; Ortega, V.E.; et al. Soluble receptor for advanced glycation end products (sRAGE) as a biomarker of COPD. Respir. Res. 2021, 22, 127. [Google Scholar] [CrossRef]
- Sellegounder, D.; Zafari, P.; Rajabinejad, M.; Taghadosi, M.; Kapahi, P. Advanced glycation end products (AGEs) and its receptor, RAGE, modulate age-dependent COVID-19 morbidity and mortality. A review and hypothesis. Int. Immunopharmacol. 2021, 98, 107806. [Google Scholar] [CrossRef]
- Saputra, G.N.R.; Yudhawati, R.; Fitriah, M. Association of soluble receptor for advanced glycation end-products (sRAGE) serum on COVID-19 severity: A cross-sectional study. Ann. Med. Surg. 2022, 74, 103303. [Google Scholar] [CrossRef]
- Zhang, X.; Li, D.; Sun, R.; Hu, X.; Song, Z.; Ni, X.; Zhu, H.; Guo, T.; Qin, C.; Xiao, R.-P. sRAGE alleviates SARS-CoV-2-induced pneumonia in hamster. Signal Transduct. Target. Ther. 2022, 7, 36. [Google Scholar] [CrossRef]
- Jessop, F.; Schwarz, B.; Scott, D.; Roberts, L.M.; Bohrnsen, E.; Hoidal, J.R.; Bosio, C.M. Impairing RAGE signaling promotes survival and limits disease pathogenesis following SARS-CoV-2 infection in mice. JCI Insight 2022, 7, e155896. [Google Scholar] [CrossRef]
- Sourris, K.C.; Watson, A.; Jandeleit-Dahm, K. Inhibitors of Advanced Glycation End Product (AGE) Formation and Accumulation. Handb. Exp. Pharmacol. 2021, 264, 395–423. [Google Scholar]
- Nagai, R.; Shirakawa, J.-I.; Ohno, R.-I.; Moroishi, N.; Nagai, M. Inhibition of AGEs formation by natural products. Amino Acids 2014, 46, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Engelen, L.; Stehouwer, C.D.A.; Schalkwijk, C.G. Current therapeutic interventions in the glycation pathway: Evidence from clinical studies. Diabetes Obes. Metab. 2013, 15, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Milne, R.; Brownstein, S. Advanced glycation end products and diabetic retinopathy. Amino Acids 2013, 44, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.; Gardiner, T.A.; Anderson, N.L.; Canning, P.; Frizzell, N.; Duffy, N.; Boyle, C.; Januszewski, A.S.; Chachich, M.; Baynes, J.W.; et al. The AGE inhibitor pyridoxamine inhibits development of retinopathy in experimental diabetes. Diabetes 2002, 51, 2826–2832. [Google Scholar] [CrossRef]
- Menini, S.; Iacobini, C.; Fantauzzi, C.B.; Pugliese, G. L-carnosine and its Derivatives as New Therapeutic Agents for the Prevention and Treatment of Vascular Complications of Diabetes. Curr. Med. Chem. 2020, 27, 1744–1763. [Google Scholar] [CrossRef]
- Lavilla, C., Jr.; Billacura, M.P.; Hanna, K.; Boocock, D.J.; Coveney, C.; Miles, A.K.; Foulds, G.A.; Murphy, A.; Tan, A.; Jackisch, L.; et al. Carnosine protects stimulus-secretion coupling through prevention of protein carbonyl adduction events in cells under metabolic stress. Free. Radic. Biol. Med. 2021, 175, 65–79. [Google Scholar] [CrossRef]
- Behl, T.; Gupta, A.; Chigurupati, S.; Singh, S.; Sehgal, A.; Badavath, V.N.; Alhowail, A.; Mani, V.; Bhatia, S.; Al-Harrasi, A.; et al. Natural and Synthetic Agents Targeting Reactive Carbonyl Species against Metabolic Syndrome. Molecules 2022, 27, 1583. [Google Scholar] [CrossRef]
- Iacobini, C.; Menini, S.; Blasetti Fantauzzi, C.; Pesce, C.M.; Giaccari, A.; Salomone, E.; Lapolla, A.; Orioli, M.; Aldini, G.; Pugliese, G. FL-926-16, a novel bioavailable carnosinase-resistant carnosine derivative, prevents onset and stops progression of diabetic nephropathy in db/db mice. Br. J. Pharmacol. 2018, 175, 53–66. [Google Scholar] [CrossRef]
- Khalid, M.; Petroianu, G.; Adem, A. Advanced Glycation End Products and Diabetes Mellitus: Mechanisms and Perspectives. Biomolecules 2022, 12, 542. [Google Scholar] [CrossRef]
- Gogoi, M.; Karmakar, K.; Chandra, K.; Chakravortty, D. Methylglyoxal and other AGEs: AGEs: Good and bad dual role in the body. In Dietary AGEs and Their Role in Health and Disease; Taylor & Francis: Abingdon, UK, 2018; pp. 365–377. [Google Scholar] [CrossRef]
- Miyata, T.; Ishikawa, S.; Asahi, K.; Inagi, R.; Suzuki, D.; Horie, K.; Tatsumi, K.; Kurokawa, K. 2-Isopropylidenehydrazono-4-oxo-thiazolidin-5-ylacetanilide (OPB-9195) treatment inhibits the development of intimal thickening after balloon injury of rat carotid artery: Role of glycoxidation and lipoxidation reactions in vascular tissue damage. FEBS Lett. 1999, 445, 202–206. [Google Scholar] [CrossRef]
- Friedman, E.A. Advanced glycosylated end products and hyperglycemia in the pathogenesis of diabetic complications. Diabetes Care 1999, 22 (Suppl. 2), B65–B71. [Google Scholar] [PubMed]
- Freedman, B.I.; Wuerth, J.P.; Cartwright, K.; Bain, R.P.; Dippe, S.; Hershon, K.; Mooradian, A.D.; Spinowitz, B.S. Design and baseline characteristics for the aminoguanidine Clinical Trial in Overt Type 2 Diabetic Nephropathy (ACTION II). Control. Clin. Trials 1999, 20, 493–510. [Google Scholar] [CrossRef]
- Borg, D.J.; Forbes, J.M. Targeting advanced glycation with pharmaceutical agents: Where are we now? Glycoconj. J. 2016, 33, 653–670. [Google Scholar] [CrossRef] [PubMed]
- Bolton, W.K.; Cattran, D.C.; Williams, M.E.; Adler, S.G.; Appel, G.B.; Cartwright, K.; Foiles, P.G.; Freedman, B.I.; Raskin, P.; Ratner, R.E.; et al. Randomized Trial of an Inhibitor of Formation of Advanced Glycation End Products in Diabetic Nephropathy. Am. J. Nephrol. 2004, 24, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Turgut, F.; Bolton, W.K. Potential new therapeutic agents for diabetic kidney disease. Am. J. Kidney Dis. 2010, 55, 928–940. [Google Scholar] [CrossRef]
- Vasan, S.; Foiles, P.; Founds, H. Therapeutic potential of breakers of advanced glycation end product-protein crosslinks. Arch. Biochem. Biophys. 2003, 419, 89–96. [Google Scholar] [CrossRef]
- Unlucerci, Y.; Bekpinar, S.; Guerdoel, F.; Seferoglu, G. A study on the relationship between homocysteine and diabetic nephropathy in rats. Pharmacol. Res. 2002, 45, 249–252. [Google Scholar] [CrossRef]
- Tamarat, R.; Silvestre, J.-S.; Huijberts, M.; Benessiano, J.; Ebrahimian, T.G.; Duriez, M.; Wautier, M.-P.; Wautier, J.L.; Levy, B.I. Blockade of advanced glycation end-product formation restores ischemia-induced angiogenesis in diabetic mice. Proc. Natl. Acad. Sci. USA 2003, 100, 8555–8560. [Google Scholar] [CrossRef]
- Mentink, C.J.A.L.; Hendriks, M.; Levels, A.A.G.; Wolffenbuttel, B.H.R. Glucose-mediated cross-linking of collagen in rat tendon and skin. Clin. Chim. Acta 2002, 321, 69–76. [Google Scholar] [CrossRef]
- Cartledge, J.J.; Eardley, I.; Morrison, J.F.B. Advanced glycation end-products are responsible for the impairment of corpus cavernosal smooth muscle relaxation seen in diabetes. BJU Int. 2001, 87, 402–407. [Google Scholar] [CrossRef]
- Cantini, C.; Kieffer, P.; Corman, B.; Liminana, P.; Atkinson, J.; Lartaud-Ldjouadiene, I. Aminoguanidine and aortic wall mechanics, structure, and composition in aged rats. Hypertension 2001, 38, 943–948. [Google Scholar] [CrossRef] [Green Version]
- Asif, M.; Egan, J.; Vasan, S.; Jyothirmayi, G.N.; Masurekar, M.R.; Lopez, S.; Williams, C.; Torres, R.L.; Wagle, D.; Ulrich, P.; et al. An advanced glycation endproduct cross-link breaker can reverse age-related increases in myocardial stiffness. Proc. Natl. Acad. Sci. USA 2000, 97, 2809–2813. [Google Scholar] [CrossRef]
- Aronson, D. Cross-linking of glycated collagen in the pathogenesis of arterial and myocardial stiffening of aging and diabetes. J. Hypertens. 2003, 21, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.J.; Lutgers, H.L. The clinical relevance of advanced glycation end products (AGE) and recent developments in pharmaceutics to reduce AGE accumulation. Curr. Med. Chem. 2004, 11, 2767–2784. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, C.G. Therapeutic interventions in the glyc(oxid)ation pathway. Immunol. Endocr. Metab. Agents Med. Chem. 2007, 7, 57–68. [Google Scholar] [CrossRef]
- Lin, Y.-T.; Tseng, Y.-Z.; Chang, K.-C. Aminoguanidine prevents fructose-induced arterial stiffening in Wistar rats: Aortic impedance analysis. Exp. Biol. Med. 2004, 229, 1038–1045. [Google Scholar] [CrossRef]
- Chang, K.C.; Liang, J.T.; Tseng, C.D.; Wu, E.T.; Hsu, K.L.; Wu, M.S.; Lin, Y.T.; Tseng, Y.Z. Aminoguanidine prevents fructose-induced deterioration in left ventricular-arterial coupling in Wistar rats. Br. J. Pharmacol. 2007, 151, 341–346. [Google Scholar] [CrossRef]
- Aronson, D. Pharmacological prevention of cardiovascular aging—Targeting the Maillard reaction. Br. J. Pharmacol. 2004, 142, 1055–1058. [Google Scholar] [CrossRef]
- Baynes, J.W.; Thorpe, S.R. Glycoxidation and lipoxidation in atherogenesis. Free. Radic. Biol. Med. 2000, 28, 1708–1716. [Google Scholar] [CrossRef]
- Degenhardt, T.P.; Alderson, N.L.; Arrington, D.D.; Beattie, R.J.; Basgen, J.M.; Steffes, M.W.; Thorpe, S.R.; Baynes, J.W. Pyridoxamine inhibits early renal disease and dyslipidemia in the streptozotocin-diabetic rat. Kidney Int. 2002, 61, 939–950. [Google Scholar] [CrossRef]
- Huijberts, M.S.P.; Wolffenbuttel, B.H.R.; Struijker Boudier, H.A.J.; Crijns, F.R.L.; Nieuwenhuijzen Kruseman, A.C.; Poitevin, P.; Lévy, B.I. Aminoguanidine treatment increases elasticity and decreases fluid filtration of large arteries from diabetic rats. J. Clin. Investig. 1993, 92, 1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggiero-Lopez, D.; Lecomte, M.; Moinet, G.; Patereau, G.; Lagarde, M.; Wiernsperger, N. Reaction of metformin with dicarbonyl compounds. Possible implication in the inhibition of advanced glycation end product formation. Biochem. Pharmacol. 1999, 58, 1765–1773. [Google Scholar] [CrossRef]
- Miyata, T.; Ueda, Y.; Yamada, Y.; Izuhara, Y.; Wada, T.; Jadoul, M.; Saito, A.; Kurokawa, K.; Van Ypersele De Strihou, C. Accumulation of carbonyls accelerates the formation of pentosidine, an advanced glycation end product: Carbonyl stress in uremia. J. Am. Soc. Nephrol. 1998, 9, 2349–2356. [Google Scholar] [CrossRef] [PubMed]
- Iacobini, C.; Vitale, M.; Haxhi, J.; Pesce, C.; Pugliese, G.; Menini, S. Food-Related Carbonyl Stress in Cardiometabolic and Cancer Risk Linked to Unhealthy Modern Diet. Nutrients 2022, 14, 1061. [Google Scholar] [CrossRef] [PubMed]
- Dimitropoulos, A.; Rosado, C.J.; Thomas, M.C. Dicarbonyl-mediated AGEing and diabetic kidney disease. J. Nephrol. 2020, 33, 909–915. [Google Scholar] [CrossRef]
- Norton, G.R.; Candy, G.; Woodiwiss, A.J. Aminoguanidine prevents the decreased myocardial compliance produced by streptozotocin-induced diabetes mellitus in rats. Circulation 1996, 93, 1905–1912. [Google Scholar] [CrossRef]
- Bodiga, V.L.; Eda, S.R.; Bodiga, S. Advanced glycation end products: Role in pathology of diabetic cardiomyopathy. Heart Fail. Rev. 2014, 19, 49–63. [Google Scholar] [CrossRef]
- Chang, K.-C.; Hsu, K.-L.; Tseng, C.-D.; Lin, Y.-D.; Cho, Y.-L.; Tseng, Y.-Z. Aminoguanidine prevents arterial stiffening and cardiac hypertrophy in streptozotocin-induced diabetes in rats. Br. J. Pharmacol. 2006, 147, 944–950. [Google Scholar] [CrossRef]
- Chang, K.-C.; Tseng, C.-D.; Wu, M.-S.; Liang, J.-T.; Tsai, M.-S.; Cho, Y.-L.; Tseng, Y.-Z. Aminoguanidine prevents arterial stiffening in a new rat model of type 2 diabetes. Eur. J. Clin. Investig. 2006, 36, 528–535. [Google Scholar] [CrossRef]
- Abraham, P.; Rabi, S.; Selvakumar, D. Protective effect of aminoguanidine against oxidative stress and bladder injury in cyclophosphamide-induced hemorrhagic cystitis in rat. Cell Biochem. Funct. 2009, 27, 56–62. [Google Scholar] [CrossRef]
- Chetyrkin, S.V.; Mathis, M.E.; Ham, A.-J.L.; Hachey, D.L.; Hudson, B.G.; Voziyan, P.A. Propagation of protein glycation damage involves modification of tryptophan residues via reactive oxygen species: Inhibition by pyridoxamine. Free. Radic. Biol. Med. 2008, 44, 1276–1285. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.E. New potential agents in treating diabetic kidney disease: The fourth act. Drugs 2006, 66, 2287–2298. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Yakupu, A.; Guan, H.; Dong, J.; Liu, Y.; Song, F.; Tang, J.; Tian, M.; Niu, Y.; Lu, S. Pyridoxamine ameliorates methylglyoxal-induced macrophage dysfunction to facilitate tissue repair in diabetic wounds. Int. Wound J. 2022, 19, 52–63. [Google Scholar] [CrossRef]
- Williams, M.E.; Bolton, W.K.; Khalifah, R.G.; Degenhardt, T.P.; Schotzinger, R.J.; McGill, J.B. Effects of Pyridoxamine in Combined Phase 2 Studies of Patients with Type 1 and Type 2 Diabetes and Overt Nephropathy. Am. J. Nephrol. 2007, 27, 605–614. [Google Scholar] [CrossRef]
- Ahmad, S.; Shahab, U.; Baig, M.H.; Khan, M.S.; Khan, M.S.; Srivastava, A.K.; Saeed, M. Inhibitory effect of metformin and pyridoxamine in the formation of early, intermediate and advanced glycation end-products. PLoS ONE 2013, 8, e72128. [Google Scholar] [CrossRef]
- Garg, S.; Syngle, A.; Vohra, K. Efficacy and tolerability of advanced glycation end-products inhibitor in osteoarthritis: A randomized, double-blind, placebo-controlled study. Clin. J. Pain 2013, 29, 717–724. [Google Scholar] [CrossRef]
- Syngle, A.; Vohra, K.; Garg, N.; Kaur, L.; Chand, P. Advanced glycation end-products inhibition improves endothelial dysfunction in rheumatoid arthritis. Int. J. Rheum. Dis. 2012, 15, 45–55. [Google Scholar] [CrossRef]
- Ooi, H.; Nasu, R.; Furukawa, A.; Takeuchi, M.; Koriyama, Y. Pyridoxamine and aminoguanidine attenuate the abnormal aggregation of β-tubulin and suppression of neurite outgrowth by glyceraldehyde-derived toxic advanced glycation end-products. Front. Pharmacol. 2022, 13, 921611. [Google Scholar] [CrossRef]
- Almeida, F.; Santos-Silva, D.; Rodrigues, T.; Matafome, P.; Crisostomo, J.; Sena, C.; Goncalves, L.; Seica, R. Pyridoxamine Reverts Methylglyoxal-induced Impairment of Survival Pathways During Heart Ischemia. Cardiovasc. Ther. 2013, 31, e79. [Google Scholar] [CrossRef]
- Kamphuis, J.B.J.; Reber, L.L.; Eutamene, H.; Theodorou, V. Increased fermentable carbohydrate intake alters colonic mucus barrier function through glycation processes and increased mast cell counts. FASEB J. 2022, 36, e22297. [Google Scholar] [CrossRef]
- Kim, J.; Kim, N.H.; Sohn, E.; Kim, C.-S.; Kim, J.S. Methylglyoxal induces cellular damage by increasing argpyrimidine accumulation and oxidative DNA damage in human lens epithelial cells. Biochem. Biophys. Res. Commun. 2010, 391, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Dal Bello, F.; Cento, A.S.; Gaens, K.; Collotta, D.; Aragno, M.; Medana, C.; Collino, M.; Wouters, K.; Schalkwijk, C.G. Altered hepatic sphingolipid metabolism in insulin resistant mice: Role of advanced glycation endproducts. Free Radic. Biol. Med. 2021, 169, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Alshanwani, A.R.; Hagar, H.; Shaheen, S.; Alhusaini, A.M.; Arafah, M.M.; Faddah, L.M.; Alharbi, F.M.B.; Sharma, A.K.; Fayed, A.; Badr, A.M. A promising antifibrotic drug, pyridoxamine attenuates thioacetamide-induced liver fibrosis by combating oxidative stress, advanced glycation end products, and balancing matrix metalloproteinases. Eur. J. Pharmacol. 2022, 923, 174910. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Khan, S.; Almatroudi, A.; Khan, A.A.; Alsahli, M.A.; Almatroodi, S.A.; Rahmani, A.H. A review on mechanism of inhibition of advanced glycation end products formation by plant derived polyphenolic compounds. Mol. Biol. Rep. 2021, 48, 787–805. [Google Scholar] [CrossRef]
- Reddy, V.P.; Zhu, X.; Perry, G.; Smith, M.A. Oxidative Stress in Diabetes and Alzheimer’s Disease. J. Alzheimer’s Dis. 2009, 16, 763–774. [Google Scholar] [CrossRef]
- Reddy, V.P.; Aryal, P.; Robinson, S.; Rafiu, R.; Obrenovich, M.; Perry, G. Polyphenols in Alzheimer’s disease and in the gut-brain axis. Microorganisms 2020, 8, 199. [Google Scholar] [CrossRef]
- Frizzell, N.; Baynes, J.W. Chelation therapy for the management of diabetic complications: A hypothesis and a proposal for clinical laboratory assessment of metal ion homeostasis in plasma. Clin. Chem. Lab. Med. 2014, 52, 69–75. [Google Scholar] [CrossRef]
- Lamas, G.A.; Goertz, C.; Boineau, R.; Mark, D.B.; Rozema, T.; Nahin, R.L.; Drisko, J.A.; Lee, K.L. Design of the Trial to Assess Chelation Therapy (TACT). Am. Heart J. 2012, 163, 7–12. [Google Scholar] [CrossRef]
- Escolar, E.; Lamas, G.A.; Mark, D.B.; Boineau, R.; Goertz, C.; Rosenberg, Y.; Nahin, R.L.; Ouyang, P.; Rozema, T.; Magaziner, A.; et al. The effect of an EDTA-based chelation regimen on patients with diabetes mellitus and prior myocardial infarction in the Trial to Assess Chelation Therapy (TACT). Circ. Cardiovasc. Qual. Outcomes 2014, 7, 15–24. [Google Scholar] [CrossRef]
- Ujueta, F.; Arenas, I.A.; Escolar, E.; Diaz, D.; Boineau, R.; Mark, D.B.; Golden, P.; Lindblad, L.; Kim, H.; Lee, K.L.; et al. The effect of EDTA-based chelation on patients with diabetes and peripheral artery disease in the Trial to Assess Chelation Therapy (TACT). J. Diabetes Complicat. 2019, 33, 490–494. [Google Scholar] [CrossRef]
- Islam, F.; Nafady, M.H.; Islam, R.M.; Saha, S.; Rashid, S.; Akter, A.; Or-Rashid, H.-M.; Akhtar, M.F.; Perveen, A.; Ashraf, G.M.; et al. Resveratrol and neuroprotection: An insight into prospective therapeutic approaches against Alzheimer′s disease from bench to bedside. Mol. Neurobiol. 2022, 59, 4384–4404. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.F.; Li, N.; Wang, Q.; Cheng, X.J.; Li, X.M.; Liu, T.T. Resveratrol decreases the insoluble Aβ1-42 level in hippocampus and protects the integrity of the blood-brain barrier in AD rats. Neuroscience 2015, 310, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, K.; Ikeda, K.; Yamori, Y. Resveratrol inhibits ages-induced proliferation and collagen synthesis activity in vascular smooth muscle cells from stroke-prone spontaneously hypertensive rats. Biochem. Biophys. Res. Commun. 2000, 274, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Maleki, V.; Foroumandi, E.; Hajizadeh-Sharafabad, F.; Kheirouri, S.; Alizadeh, M. The effect of resveratrol on advanced glycation end products in diabetes mellitus: A systematic review. Arch. Physiol. Biochem. 2022, 128, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Ma, H.; Frost, L.; Yuan, T.; Dain, J.A.; Seeram, N.P. Pomegranate phenolics inhibit formation of advanced glycation endproducts by scavenging reactive carbonyl species. Food Funct. 2014, 5, 2996–3004. [Google Scholar] [CrossRef]
- Vasan, S.; Zhang, X.; Zhang, X.; Kapurniotu, A.; Bernhagen, J.; Teichberg, S.; Basgen, J.; Wagle, D.; Shih, D. An agent cleaving glucose-derived protein crosslinks in vitro and in vivo. Nature 1996, 382, 275–278. [Google Scholar] [CrossRef]
- Schwedler, S.B.; Verbeke, P.; Bakala, H.; Weiss, M.F.; Vilar, J.; Depreux, P.; Fourmaintraux, E.; Striker, L.J.; Striker, G.E. N-Phenacylthiazolium bromide decreases renal and increases urinary advanced glycation end products excretion without ameliorating diabetic nephropathy in C57BL/6 mice. Diabetes Obes. Metab. 2001, 3, 230–239. [Google Scholar] [CrossRef]
- Cooper, M.E.; Thallas, V.; Forbes, J.; Scalbert, E.; Sastra, S.; Darby, I.; Soulis, T. The cross-link breaker, N-phenacylthiazolium bromide prevents vascular advanced glycation end-product accumulation. Diabetologia 2000, 43, 660–664. [Google Scholar]
- Yang, S.; Litchfield, J.E.; Baynes, J.W. AGE-breakers cleave model compounds, but do not break Maillard crosslinks in skin and tail collagen from diabetic rats. Arch. Biochem. Biophys. 2003, 412, 42–46. [Google Scholar] [CrossRef]
- Kalousova, M.; Zima, T.; Tesar, V.; Stipek, S.; Sulkova, S. Advanced glycation end products in clinical nephrology. Kidney Blood Press. Res. 2004, 27, 18–28. [Google Scholar]
- Chang, P.-C.; Tsai, S.-C.; Chong, L.Y.; Kao, M.-J. N-phenacylthiazolium bromide inhibits the advanced glycation end product (AGE)-AGE receptor axis to modulate experimental periodontitis in rats. J. Periodontol. 2014, 85, e268. [Google Scholar] [CrossRef] [PubMed]
- Little, W.C.; Zile, M.R.; Kitzman, D.W.; Hundley, W.G.; O’Brien, T.X.; DeGroof, R.C. The Effect of Alagebrium Chloride (ALT-711), a Novel Glucose Cross-Link Breaker, in the Treatment of Elderly Patients with Diastolic Heart Failure. J. Card. Fail. 2005, 11, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Toprak, C.; Yigitaslan, S. Alagebrium and complications of diabetes mellitus. Eurasian J. Med. 2019, 51, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Pathak, P.; Gupta, R.; Chaudhari, A.; Shiwalkar, A.; Dubey, A.; Mandhare, A.B.; Gupta, R.C.; Joshi, D.; Chauthaiwale, V. TRC4149 a novel advanced glycation end product breaker improves hemodynamic status in diabetic spontaneously hypertensive rats. Eur. J. Med. Res. 2008, 13, 388–398. [Google Scholar]
- Joshi, D.; Gupta, R.; Dubey, A.; Shiwalkar, A.; Pathak, P.; Gupta, R.C.; Chauthaiwale, V.; Dutt, C. TRC4186, a Novel AGE-breaker, Improves Diabetic Cardiomyopathy and Nephropathy in Ob-ZSF1 Model of Type 2 Diabetes. J. Cardiovasc. Pharmacol. 2009, 54, 72–81. [Google Scholar] [CrossRef]
- Chandra, K.P.; Shiwalkar, A.; Kotecha, J.; Thakkar, P.; Srivastava, A.; Chauthaiwale, V.; Sharma, S.K.; Cross, M.R.; Dutt, C. Phase I clinical studies of the advanced glycation end-product (AGE)-breaker TRC4186: Safety, tolerability and pharmacokinetics in healthy subjects. Clin. Drug Investig. 2009, 29, 559–575. [Google Scholar] [CrossRef]
- Kim, T.; Spiegel, D.A. The Unique Reactivity of N-Phenacyl-Derived Thiazolium Salts Toward α-Dicarbonyl Compounds. Rejuvenation Res. 2013, 16, 43–50. [Google Scholar] [CrossRef]
- Nagai, R.; Murray, D.B.; Metz, T.O.; Baynes, J.W. Chelation: A fundamental mechanism of action of AGE inhibitors, AGE breakers, and other inhibitors of diabetes complications. Diabetes 2012, 61, 549–559. [Google Scholar] [CrossRef]
- Khurana, R.; Coleman, C.; Ionescu-Zanetti, C.; Carter, S.A.; Krishna, V.; Grover, R.K.; Roy, R.; Singh, S. Mechanism of thioflavin T binding to amyloid fibrils. J. Struct. Biol. 2005, 151, 229–238. [Google Scholar] [CrossRef]
- Biancalana, M.; Koide, S. Molecular mechanism of thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta Proteins Proteom. 2010, 1804, 1405–1412. [Google Scholar] [CrossRef]
- Wolffenbuttel, B.H.R.; Boulanger, C.M.; Crijns, F.R.L.; Huijberts, M.S.P.; Poitevin, P.; Swennen, G.N.M.; Vasan, S.; Egan, J.J.; Ulrich, P.; Cerami, A.; et al. Breakers of advanced glycation end products restore large artery properties in experimental diabetes. Proc. Natl. Acad. Sci. USA 1998, 95, 4630–4634. [Google Scholar] [CrossRef]
- Cheng, G.; Wang, L.L.; Long, L.; Liu, H.Y.; Cui, H.; Qu, W.S.; Li, S. Beneficial effects of C36, a novel breaker of advanced glycation endproducts cross-links, on the cardiovascular system of diabetic rats. Br. J. Pharmacol. 2007, 152, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Vaitkevicius, P.V.; Lane, M.; Spurgeon, H.; Ingram, D.K.; Roth, G.S.; Egan, J.J.; Vasan, S.; Wagle, D.R.; Ulrich, P.; Brines, M.; et al. A cross-link breaker has sustained effects on arterial and ventricular properties in older rhesus monkeys. Proc. Natl. Acad. Sci. USA 2001, 98, 1171–1175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; He, K.; Chen, W.; Cheng, X.; Cui, H.; Zhong, W.; Li, S.; Wang, L. Alagebrium (ALT-711) improves the anti-hypertensive efficacy of nifedipine in diabetic-hypertensive rats. Hypertens. Res. 2014, 37, 901–907. [Google Scholar] [CrossRef]
- Oudegeest-Sander, M.H.; Rikkert, M.G.M.O.; Smits, P.; Thijssen, D.H.J.; van Dijk, A.P.J.; Levine, B.D.; Hopman, M.T.E. The effect of an advanced glycation end-product crosslink breaker and exercise training on vascular function in older individuals: A randomized factorial design trial. Exp. Gerontol. 2013, 48, 1509–1517. [Google Scholar] [PubMed]
- Nenna, A.; Nappi, F.; Avtaar Singh, S.S.; Sutherland, F.W.; Di Domenico, F.; Chello, M.; Spadaccio, C. Pharmacologic Approaches Against Advanced Glycation End Products (AGEs) in Diabetic Cardiovascular Disease. Res. Cardiovasc. Med. 2015, 4, e26949. [Google Scholar]
- Hartog, J.W.L.; Willemsen, S.; van Veldhuisen, D.J.; Posma, J.L.; van Wijk, L.M.; Hummel, Y.M.; Hillege, H.L.; Voors, A.A. Effects of alagebrium, an advanced glycation endproduct breaker, on exercise tolerance and cardiac function in patients with chronic heart failure. Eur. J. Heart Fail. 2011, 13, 899–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reddy, V.P.; Aryal, P.; Darkwah, E.K. Advanced Glycation End Products in Health and Disease. Microorganisms 2022, 10, 1848. https://doi.org/10.3390/microorganisms10091848
Reddy VP, Aryal P, Darkwah EK. Advanced Glycation End Products in Health and Disease. Microorganisms. 2022; 10(9):1848. https://doi.org/10.3390/microorganisms10091848
Chicago/Turabian StyleReddy, V. Prakash, Puspa Aryal, and Emmanuel K. Darkwah. 2022. "Advanced Glycation End Products in Health and Disease" Microorganisms 10, no. 9: 1848. https://doi.org/10.3390/microorganisms10091848
APA StyleReddy, V. P., Aryal, P., & Darkwah, E. K. (2022). Advanced Glycation End Products in Health and Disease. Microorganisms, 10(9), 1848. https://doi.org/10.3390/microorganisms10091848