1. Introduction
Salmonellosis, one of the primary causes of foodborne infections resulting from gram-negative enteropathogenic bacteria
Salmonella spp., is a global threat to human health [
1]. Typhoidal
Salmonella causes enteric fever in humans, whereas non-typhoidal
Salmonella (NTS) results in acute/chronic gastroenteritis. Annually, it is estimated that NTS is responsible for ~93.8 million infections and ~155,000 deaths [
2].
NTS infections cause diarrhoea and a non-specific febrile illness that is clinically indistinguishable from other febrile illnesses [
3].
Salmonella enterica subspecies
enterica has more than 2600 serovars according to unique somatic (O) and flagellar (H) antigenic formulae [
4,
5].
S. enterica sv. Typhimurium and
S. enterica sv. Enteritidis are the main pathogens responsible for causing gastroenteritis in humans [
6,
7].
To prevent the occurrence of the main
Salmonella serovars worldwide, several prevention and control measures are adopted in farms and food processing industries. In Brazil,
Salmonella infection of flocks and transmission to poultry-derived food is a major transmission route for the pathogen.
Salmonella is routinely managed on Brazilian farms by poultry vaccination and laboratory testing (Available online:
https://www.gov.br/agricultura/pt-br/assuntos/sanidade-animal-e-vegetal/saude-animal/programas-desaude-animal/pnsa/2003_78.INconsolidada.pdf (accessed on 18 December 2022)). However, despite these measures several poultry diseases and foodborne
Salmonella outbreaks have been reported in Brazil in recent decades [
8].
Whole-genome sequencing (WGS) is useful in foodborne outbreak investigations and pathogen surveillance [
9]. Illumina short-read sequencing technology has proven to be robust for characterizing pathogens of clinical care [
10], but it is unable to resolve repetitive and GC-rich regions, thus producing unresolvable regions in the underlying genome assembly [
11]. These unresolved regions impede completion of a whole-genome structure, which is crucial to determine if some genes are co-regulated or co-transmissible, and if they are located on the chromosome or plasmids [
12]. Furthermore, the bias to identify key virulence genes during an outbreak investigation can also have negative impacts on public health assessment.
Nanopore sequencing technology can generate long reads to facilitate the completion of bacterial genome assemblies but can lack sequencing depth in some repetitive regions [
13]. However, nanopore’s long reads can span wide repetitive regions and help solve GC-rich regions, making it useful for resolving full-length genome sequences [
14]. Nanopore sequencing technology exhibits lower read accuracy than Illumina sequencing which can produce systematic errors, as a result, it has only usually been applied as a complement to short-read sequencing for bacterial genome assembly [
15]. Since the release of the MinION platform by Oxford Nanopore Technologies, nanopore chemistry, base-calling, and bioinformatic tools have been steadily improving and are now more able to produce accurate bacterial genome sequences independent of other sequencing technologies [
16].
The combination of both short reads for base-calling accuracy and long reads for structural integrity has recently been developed as a hybrid assembly approach to close whole-genome assemblies, such as those found in the Unicycler and SPAdes pipelines [
17,
18]. Unicycler was specifically developed for hybrid assembly of bacterial genomes [
18]. Unicycler generates a short-read assembly graph and then uses long-reads to build bridges to resolve all repeats in the genome, performs multiple rounds of short-read polishing and finally, it produces a complete genome assembly [
14].
In this study, a hybrid genome assembly approach using MinION and HiSeq sequencing data was used to improve the assembly parameters and gene completeness, identification of virulence and antimicrobial resistance genes (ARG), genome phylogeny and pangenome in Salmonella enterica var. Enteritidis SE3 isolated from soil at the Subaé river in Santo Amaro, Brazil, a river polluted with organic waste and heavy metals.
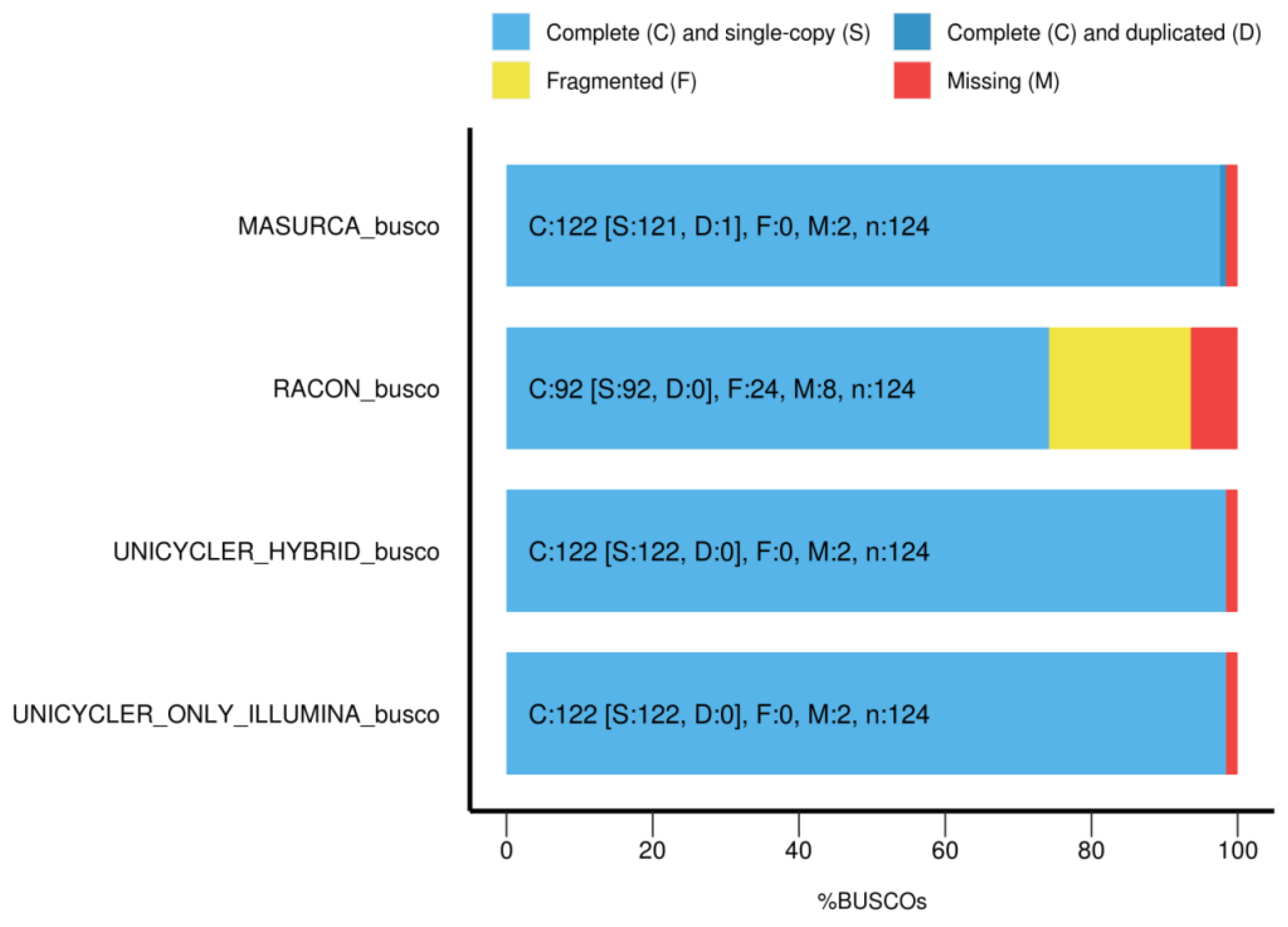
4. Discussion
Salmonella SE3 was isolated from soil at the Subaé River in Santo Amaro, Brazil, a region contaminated with heavy metals and organic waste. The genome sequence of this isolate was determined using two sequencing technologies and six different bioinformatics strategies. Hybrid assembly showed the lowest number of contigs followed by MinION-alone assembly, with hybrid genome assembly resulting in a genome of 4.73 Mb, which was similar in size to that reported (4.68 Mb) for
Salmonella enterica subsp.
enterica serovar Enteritidis str. P125109 (NC_011294.1) [
52]. However, the GC content of the assembled genome (52.16%) was more similar to
Salmonella enterica subsp. enterica serovar Enteritidis str. P125109 (NC_011294.1) (52.17%) [
52]. HiSeq assemblies have been traditionally considered the “gold standard” because MinION sequencing could introduce high numbers of errors and consequently may interfere with high-quality genome annotations due to reduced accuracy in gene prediction, producing a large number of misannotated genes [
53,
54]. However, the genome completeness of
Salmonella SE3 with non-hybrid assembly and hybrid assembly were almost identical.
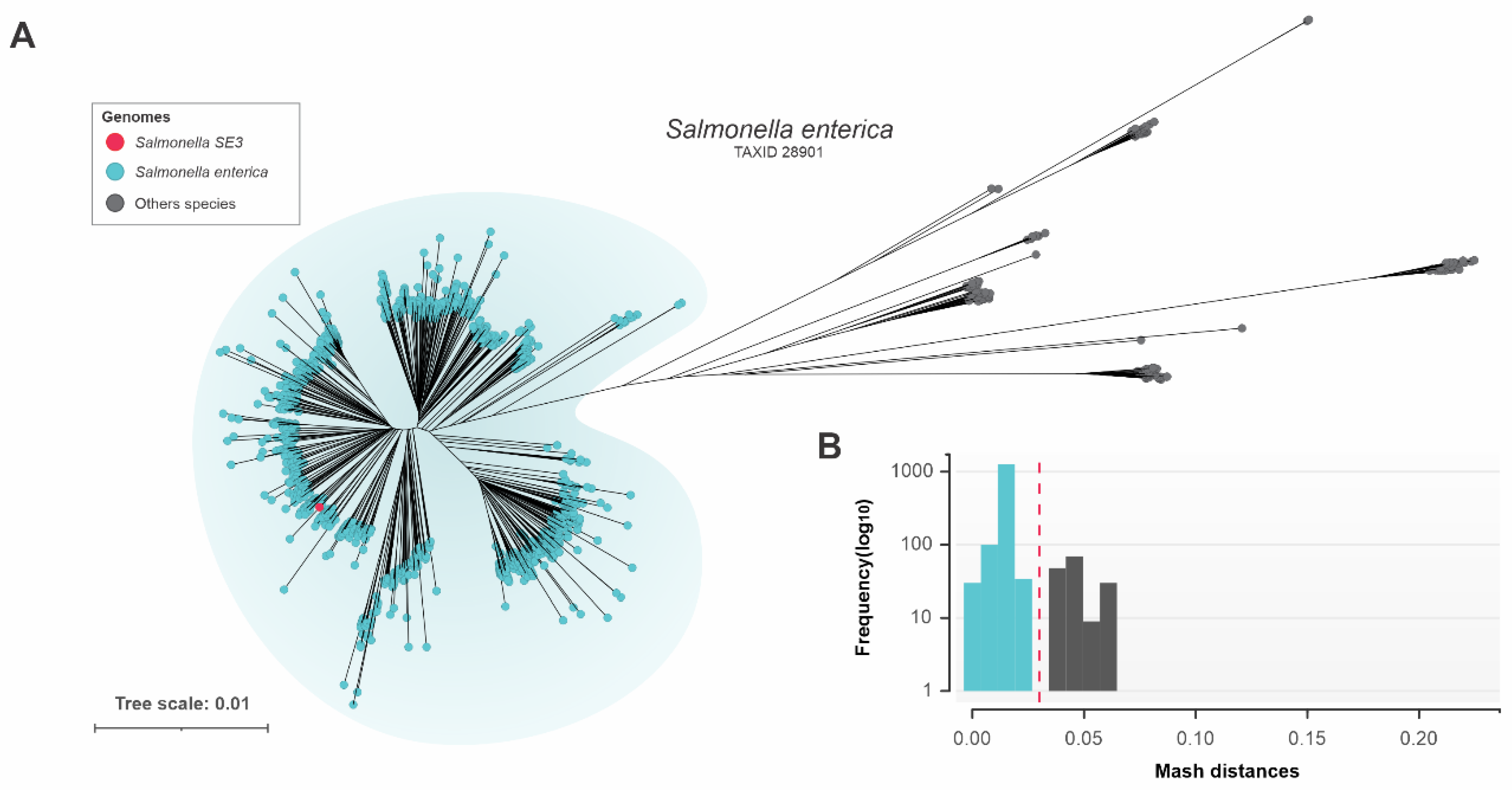
Phylogenetic analysis of the Salmonella SE3 genome revealed it was located within the properly classified cluster of S. enterica. During taxon analysis we identified 159 genomes with incorrect taxonomic classification, highlighting that it is important to confirm identity prior to undertaking phylogenetic analyses.
The pangenome analysis of
Salmonella SE3, revealed the core genome was composed of 2137 genes and the accessory genome comprised 3390 shell genes and 69,352 cloud genes. This indicates
Salmonella SE3 has an open pangenome with a diversity of unique genes. A study by Chand et al. [
55] undertook a comparative genomic analysis of 44 genome sequences, representing 17 serovars of
S. enterica, and concluded that the genus
Salmonella displays an open pangenome, comprising a reservoir of 10,775 gene families. Of these 2847 constituted the core gene families, 4657 were dispensable or accessory gene families, and 3271 strain-specific gene families. Park et al. [
56] constructed pangenomes of seven species to elucidate variations in the genetic contents of >27,000 genomes, as in our study, this work showed the pangenome of
Salmonella enterica subsp.
enterica was open. However, it is important to note that pangenome size is heavily influenced by the properties of the genomes used and variation would likely result in inconsistencies, and secondly, newly described genes are often included which results in open pangenomes [
57].
The antimicrobial resistance gene profile of
Salmonella SE3 identified genes potentially involved in resistance to aminoglycosides, fluoroquinolones, macrolides, a monobactam (
golS), nitroimidazole (
msbA), tetracycline and related drugs (
mdfA), and cephalosporins. Other studies of
Salmonella isolates from southern Brazil have also reported tetracycline (
mdfA) and aminoglycoside (
aac(6’)-Iaa) resistance genes, in addition to other genes such as
aac(3)-Iva, aph(3”)-Ib, aph(4)-Ia, aph(6)-Id, tet(34) and
tet(A) [
57,
58,
59,
60,
61]. In the United States, additional antibiotic resistance mechanisms in
S. enterica have been described [
62], such as resistance to aminoglycosides (
aadA, aadB, aacC, aphA, strAB), β-lactams (
blaCMY-2, PSE-1,
TEM-1), chloramphenicol (
cat1, cat2, cmlA, floR), inhibitors of the folate pathway (
dfr, sul), and tetracycline (
tetA, tetB, tetC, tetD, tetG, and
tetR), none of these resistance genes were detected in our study.
Ten
Salmonella pathogenic islands were identified in
Salmonella SE3 which is relatively high compared that reported for other
Salmonella isolates. A
S. enterica serovar Typhimurium isolate, ms202, from a patient in India possessed six
Salmonella pathogenicity islands: SPI-1, SPI-2, SPI-3, SPI-4, SPI-5, and SPI-11 [
63], but in our work, we did not identify SPI-4. The genes identified in SPI regions had similarity to known transporters, drug targets, and antibiotic-resistance genes, and in a subset of genomic islands, genes that facilitate the horizontal transfer of genes encoding numerous resistance and virulence factors of regions belonging to type III secretion systems (T3SS). Vilela et al. [
64] analyzed six
Salmonella Choleraesuis strains provided by the Brazilian
Salmonella reference laboratory of the Oswaldo Cruz Foundation (FIOCRUZ-RJ), which receives
Salmonella isolates from diverse isolation sources and regions of the country. Pathogenicity islands SPI-1, -2, -3, -4, -5, -9, -13, -14 and CS54 island were detected in five strains and SPI-11 in four strains. The majority of these SPI, with the exception of SPI 4 and SPI 11, were also detected in
Salmonella SE3. SPI-1 and SPI-2 are known to be involved in the invasion of intestinal epithelial cells and survival and replication within phagocytic cells, respectively, through the formation of type 3 secretion systems, SPI-5 is associated with fluid secretion and inflammatory response and SPI-3, -4, -11, -13, -14 and CS54 are associated with
Salmonella survival and adaptation to stresses within macrophages [
65].
In total, 144 potential virulence genes were identified in
Salmonella SE3. Some of these virulence genes are also found in other serovars of
Salmonella. Borah et al. [
66] investigated virulence genes in 88
Salmonella isolates recovered from humans and different species of animals. Among the 88 isolates, some virulence genes such
invA,
sipA,
sipB and
sipC were detected irrespective of the serovar, and these were also detected in
Salmonella SE3.
fepA was also present in a high percentage (64.7%) of isolates belonging to
Salmonella serovars Enteritidis, Weltervreden, Typhi, Newport, Litchfield, Idikan and Typhimurium, as well as
Salmonella SE3 and. Other virulence genes were present in varying percentages among the
Salmonella serovars studied by Borah et al. [
66] such as
sopB (86.36%),
sopE2 (62.5%),
pefA (79.54%) and
sefC (51.14%); of these genes only
sefC was not detected in
Salmonella SE3. The virulence genes identified in
Salmonella SE3 are involved in several different processes, such as the
invA gene usually codes for a protein in the inner bacterial membrane that is responsible for the invasion of intestinal cells of the host [
67,
68]. The
fepA gene encodes outer membrane receptor protein FepA, which participates in iron transport and plays a role in infection colonization in
Salmonella [
32]. T3SS-1 secretes proteins, termed effectors, across the inner and outer membranes of the bacterial cell. Some of the secreted effectors, including SipA, SipB and SipC are encoded by genes located on SPI1. The remaining effectors, including SopA, SopB, SopD, SopE and SopE2 are encoded by genes that are scattered around the
Salmonella SE3 chromosome. Upon secretion the SipB, SipC, and SipD proteins are thought to form a complex in the eukaryotic membrane that is required for translocation of the remaining effectors into the host cell cytoplasm [
69]. PefA is encoded by
Salmonella SE3 and is the plasmid-encoded fimbrial major subunit antigen of
Salmonella Typhimurium [
70].
Salmonella plasmid-encoded fimbrae have been found to mediate adhesion to mouse intestinal epithelium [
71].
The gene
arsC, encoding arsenate reductase, was found in the genome of
Salmonella SE3. Arsenate reductase is essential for arsenate resistance and transforms arsenate into arsenite which is extruded from the cell [
72,
73]. This is of interest as
Salmonella SE3 was isolated from the soil of Subaé River where heavy metal concentrations were above reference values [
74]. In addition, mussels (
Mytella charruana) gathered from the same region also contained lead, arsenic and cadmium in concentrations above reference values [
75]. Carvalho et al. [
75] also determined the quality of soils in 39 households from nearby Santo Amaro City, and the Residential Investigation Value (RIV) was exceeded by Lead (23.1% of the samples), Cadmium (7.7%), Nickel (2.6%), Zinc (25.6%), Arsenic (2.6%), and Antimony (7.7%).
Several virus defence systems were detected in
Salmonella SE3, including CRISPR-Cas type IE, CBASS type I, and RM type I and III systems. Similar antiviral systems and subtypes were identified by the PADLOC and DefenseFinder tools, except for AbiU and RM type II which were only identified by PADLOC. Most bacteria, including
Salmonella, possess multiple antiviral defence systems that protect against infection by phages and mobile genetic elements [
47].
Seven prophages were detected in the
Salmonella SE3 genome, two were intact, and five were incomplete. By comparison, in
S. enterica Typhimurium ms202 nine prophages were detected, two were intact, five were incomplete and two were questionable [
63]. Moreover,
Salmonella SE3 had not only
Salmonella prophage sequences (e.g. phage RE-2010) but also prophages annotated as belonging to closely related genera
Shigella (phage POCJ13) and
Escherichia (phage 500465-2), which may indicate horizontal gene transfer or polyvalent phages. A previous study has reported that phage populations in
S. enterica contribute to horizontal gene transfer, including virulence and virulence-related genes within the subspecies [
76,
77,
78,
79]. Further studies on
Salmonella phages may uncover the receptor-interaction mechanisms between phages and hosts which may lead to improving phage therapy as an option for the treatment or control of
Salmonella.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}