
Distinct Microbiotas Are Associated with Different Production Lines in the Cutting Room of a Swine Slaughterhouse

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Facility Structure, Sampling, Total Microbiota Harvesting, DNA Extraction, Amplicon Library Preparation, and 16S rRNA Sequencing
2.2. Sequencing Data Processing, Diversity, and Statistical Analysis
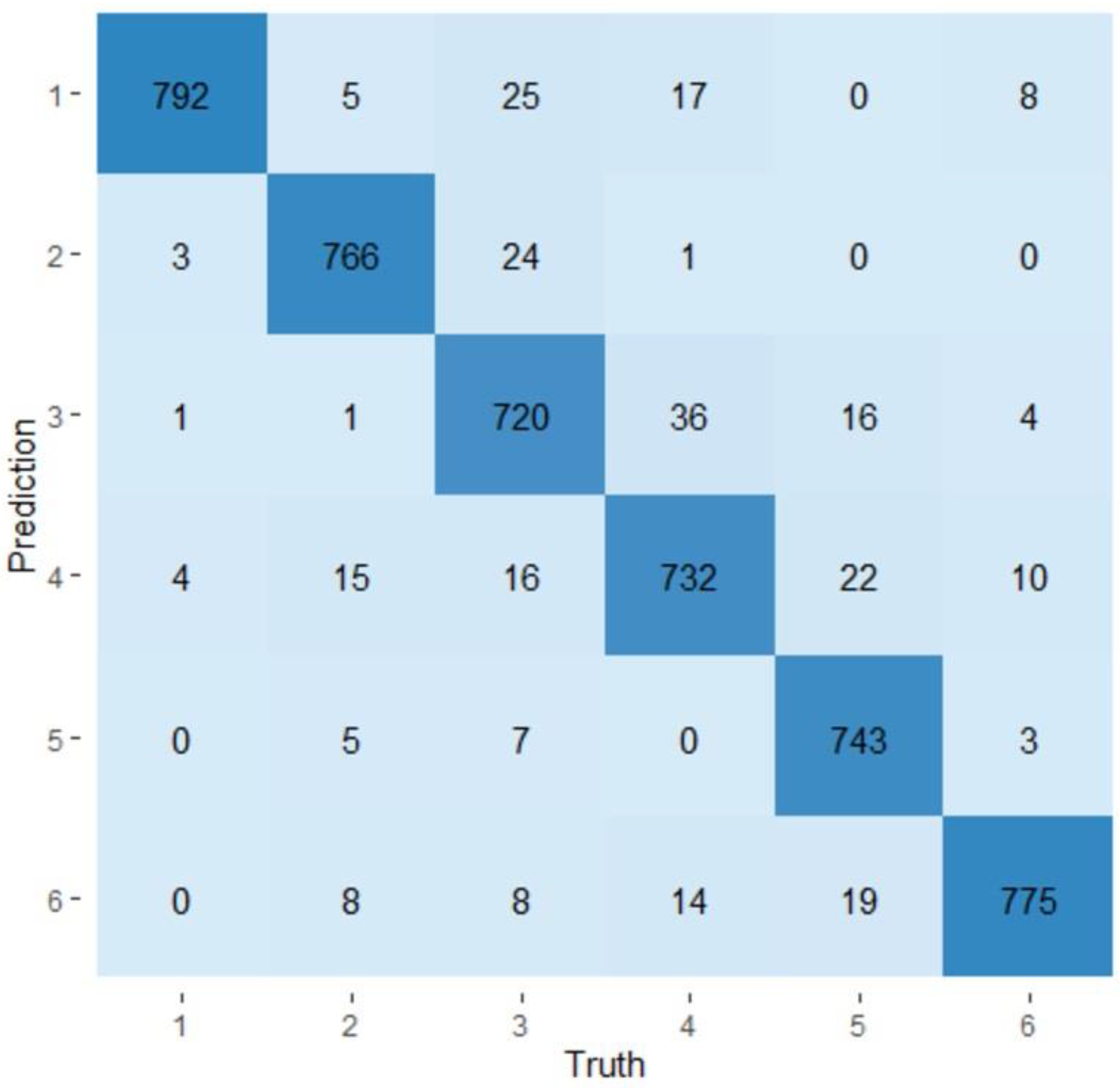
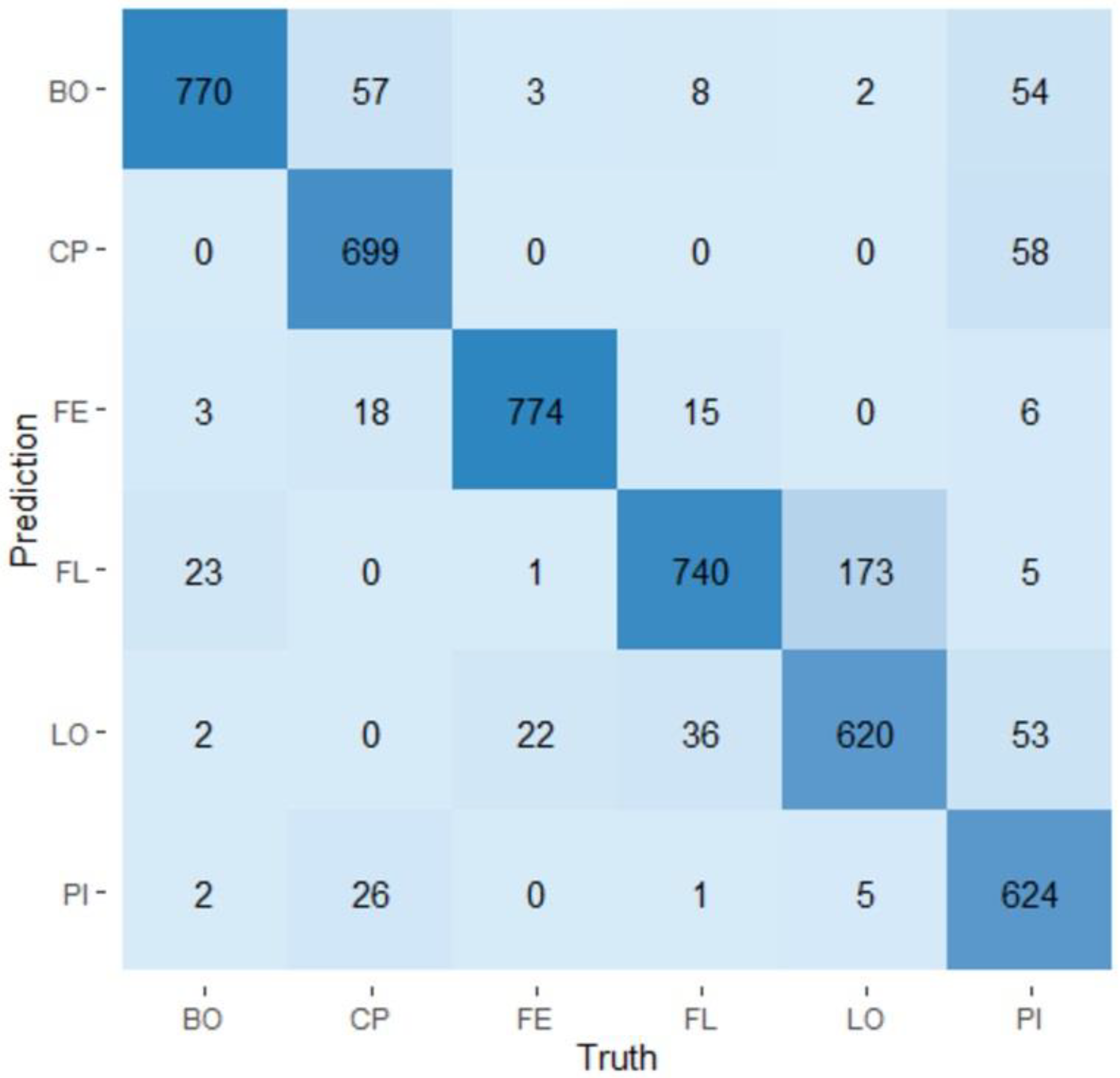
2.3. Evaluation of the Predictability of a Visit and a Production Line Using Random Forest Models
3. Results
3.1. 16S rRNA Gene Amplicon Sequencing
3.2. Production Line Surface Microbiota Description
3.3. Microbiota Diversity of Conveyor Belt Surfaces Based on Visit and on Production Line
3.3.1. Alpha Diversity
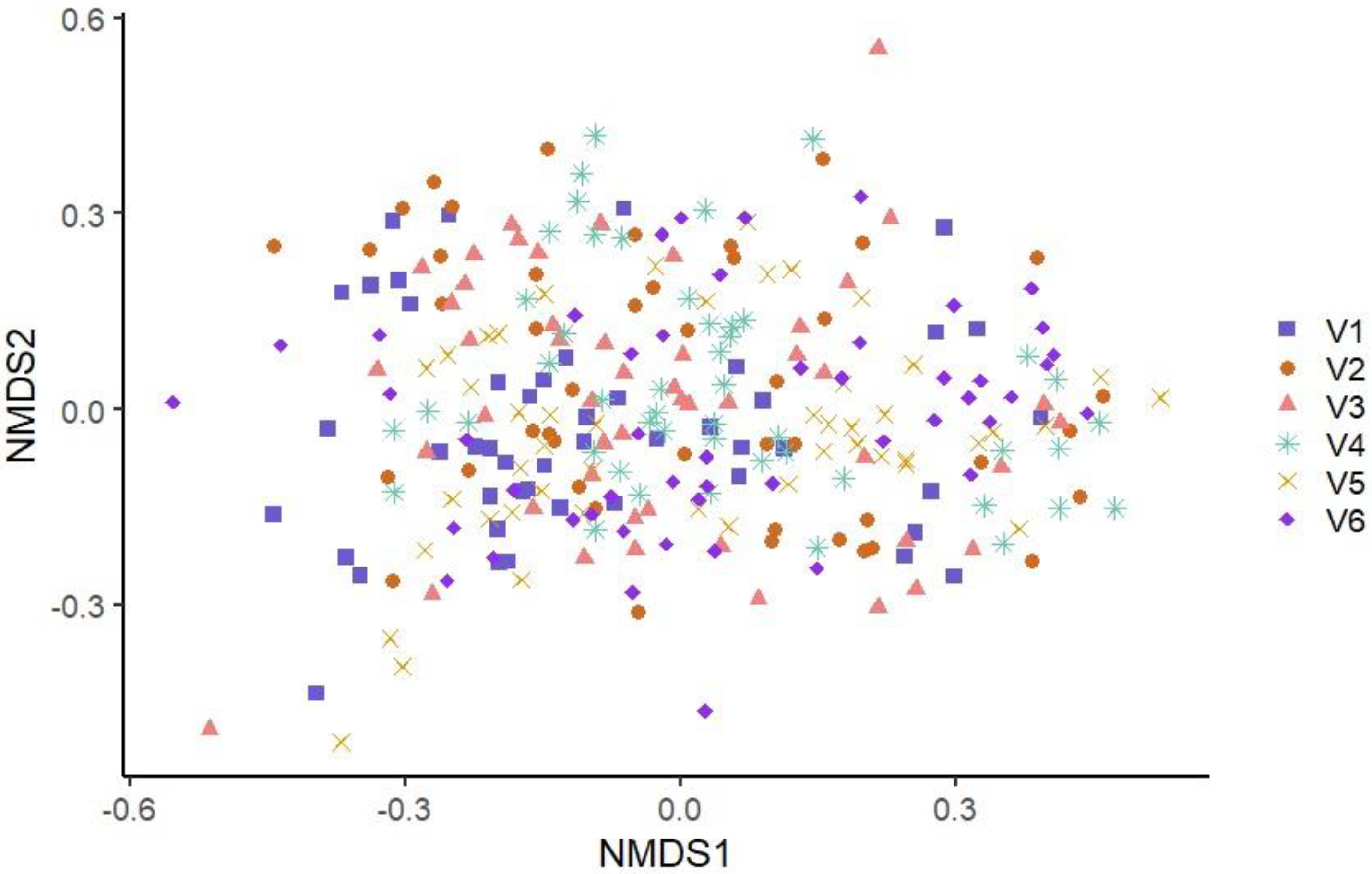
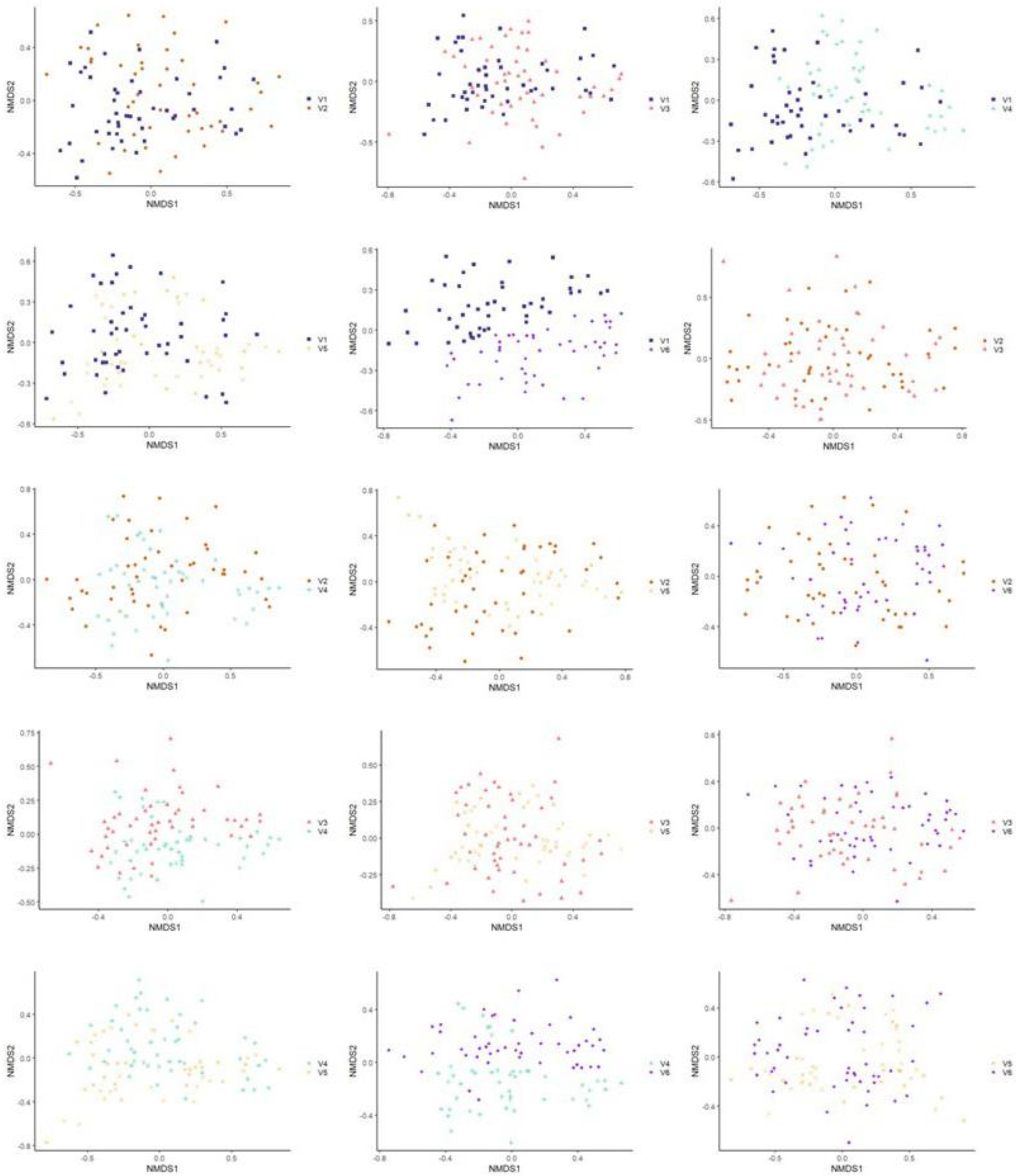
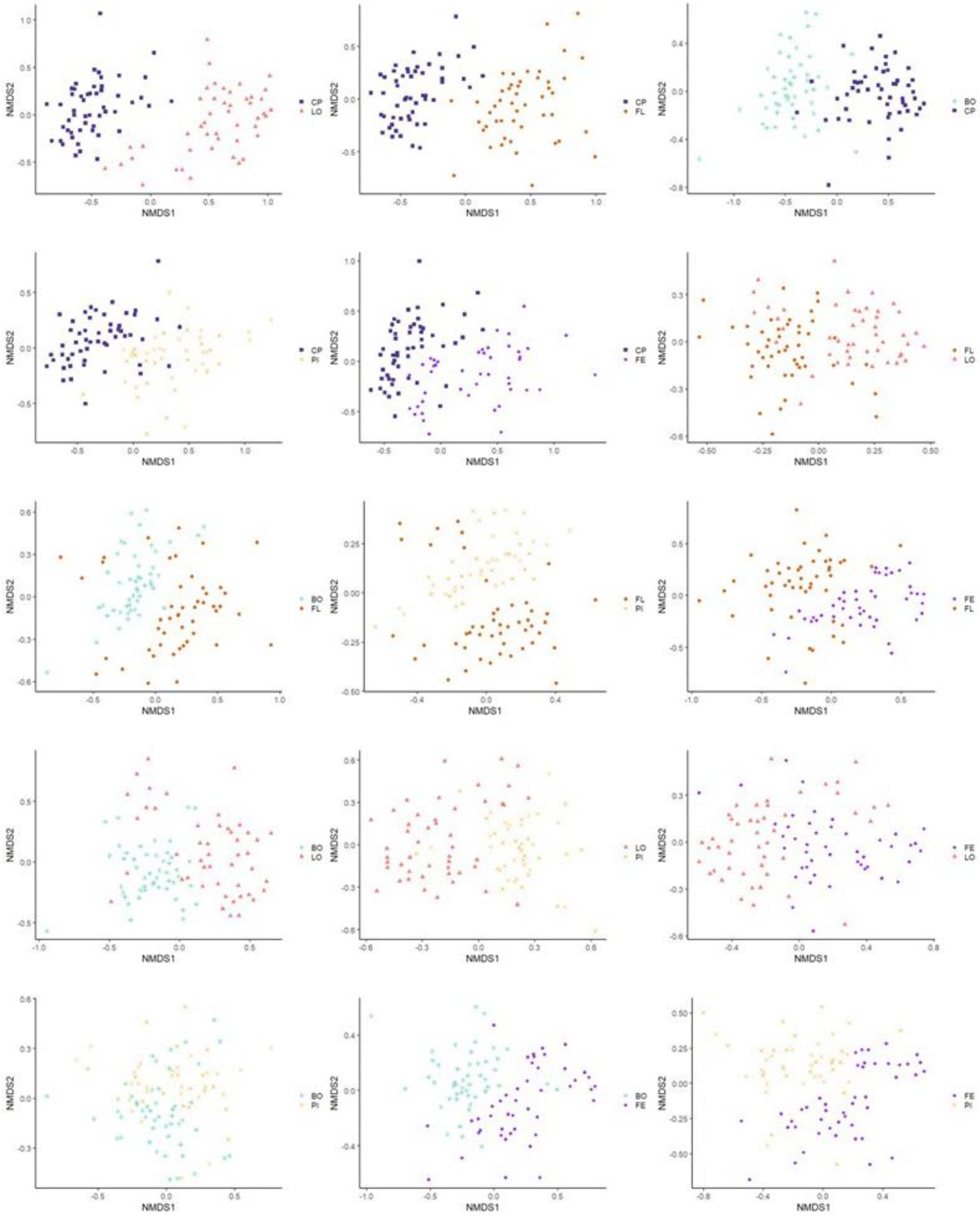
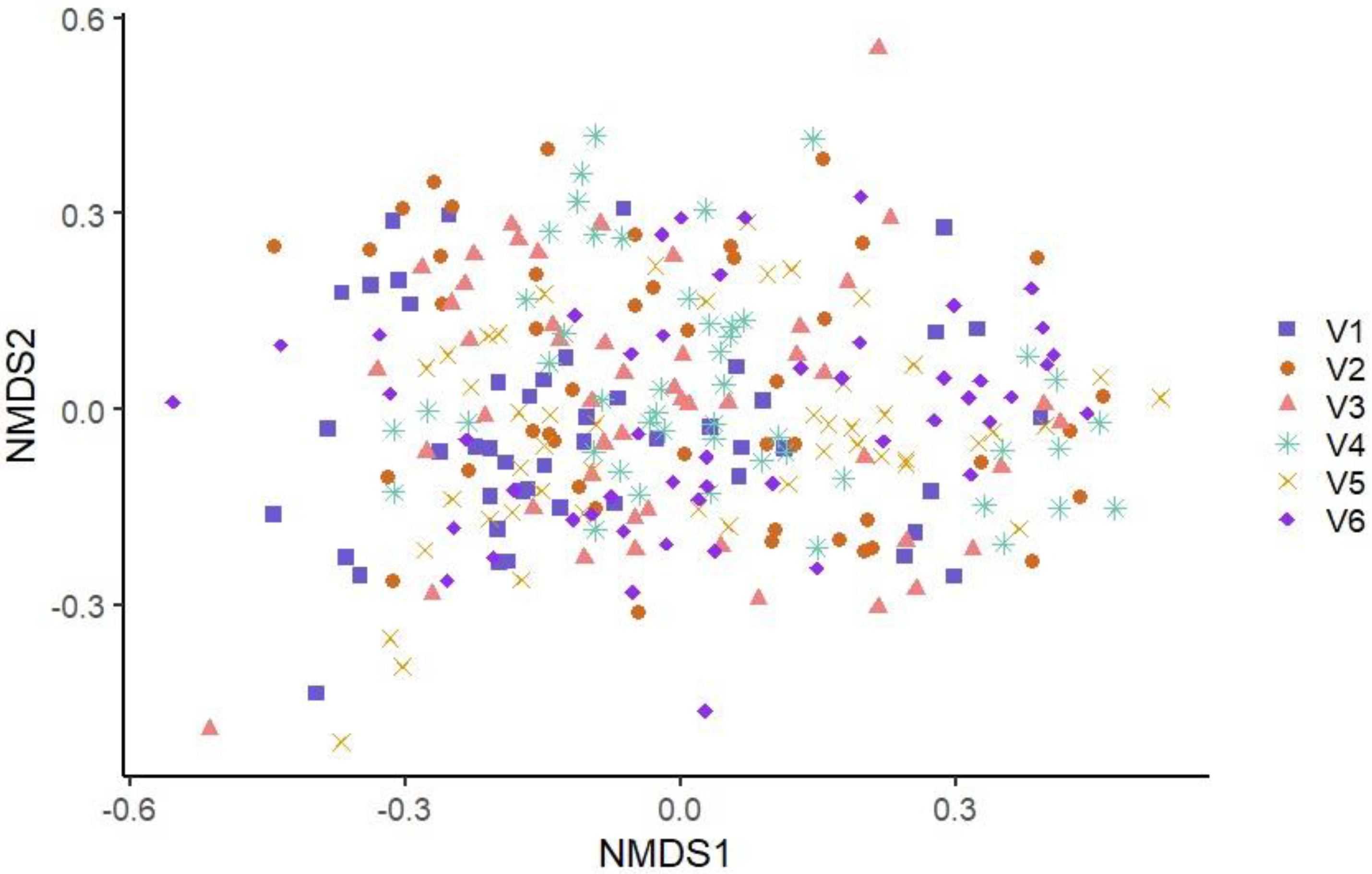
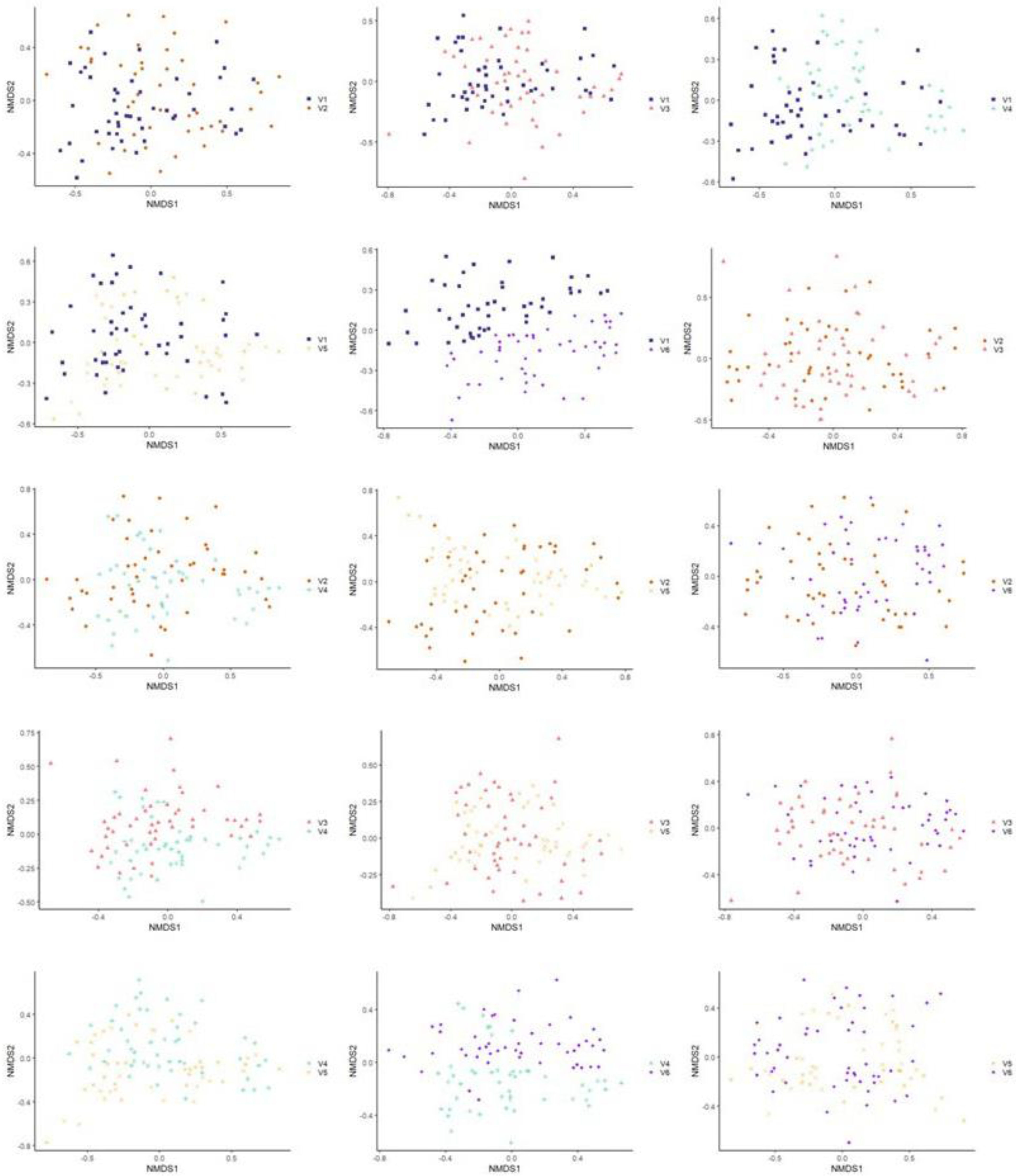
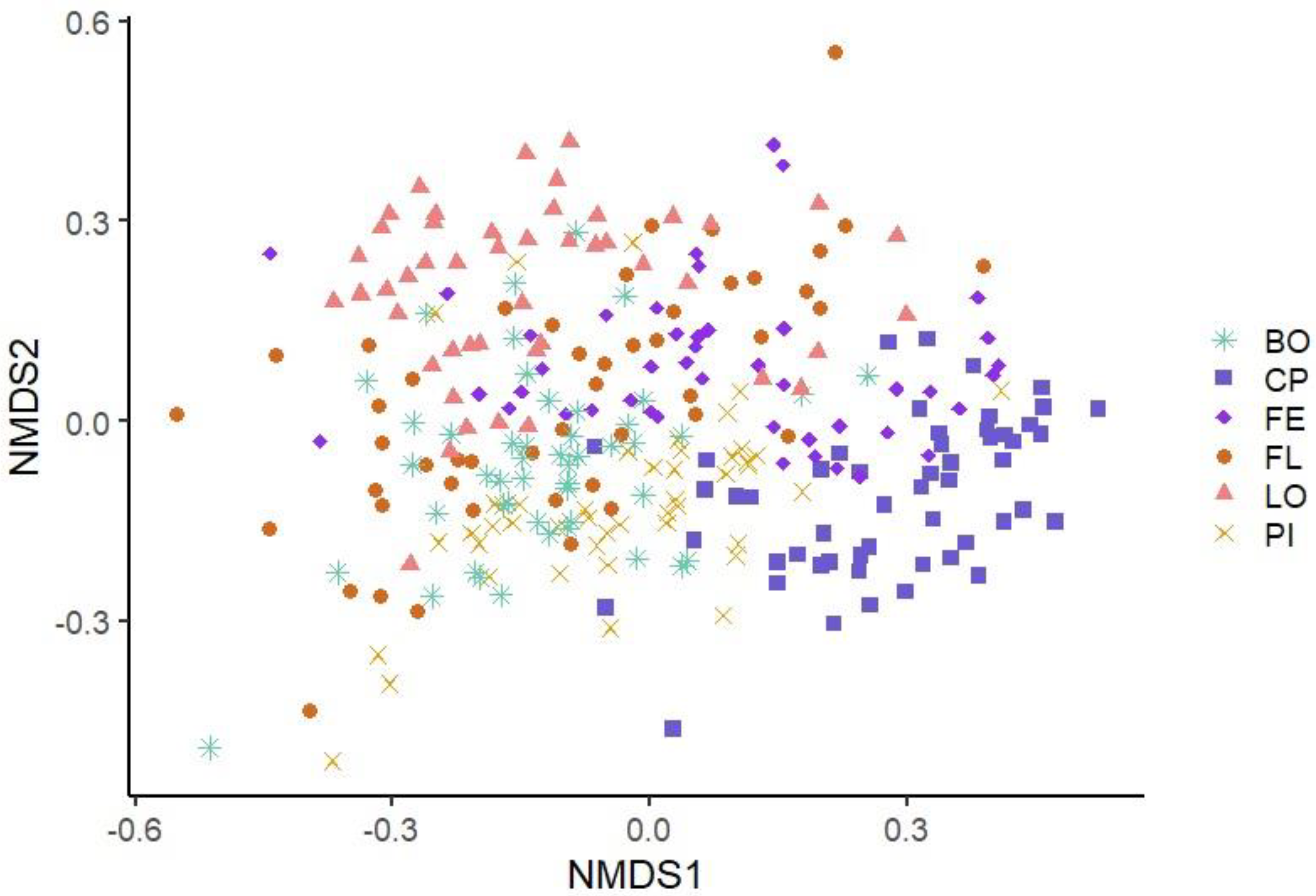
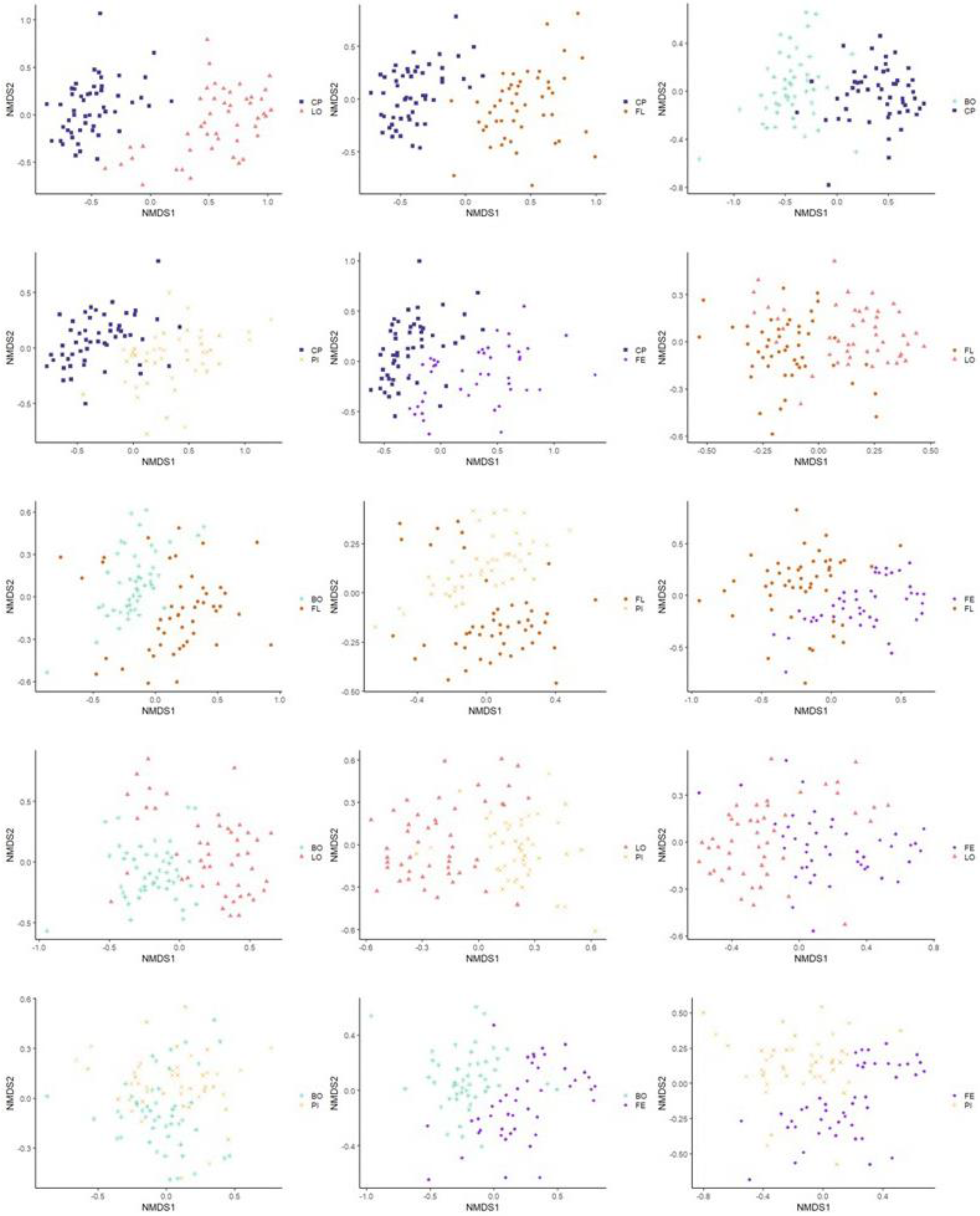
3.3.2. Beta Diversity
3.3.3. Multivariate Association with Linear Model Analysis
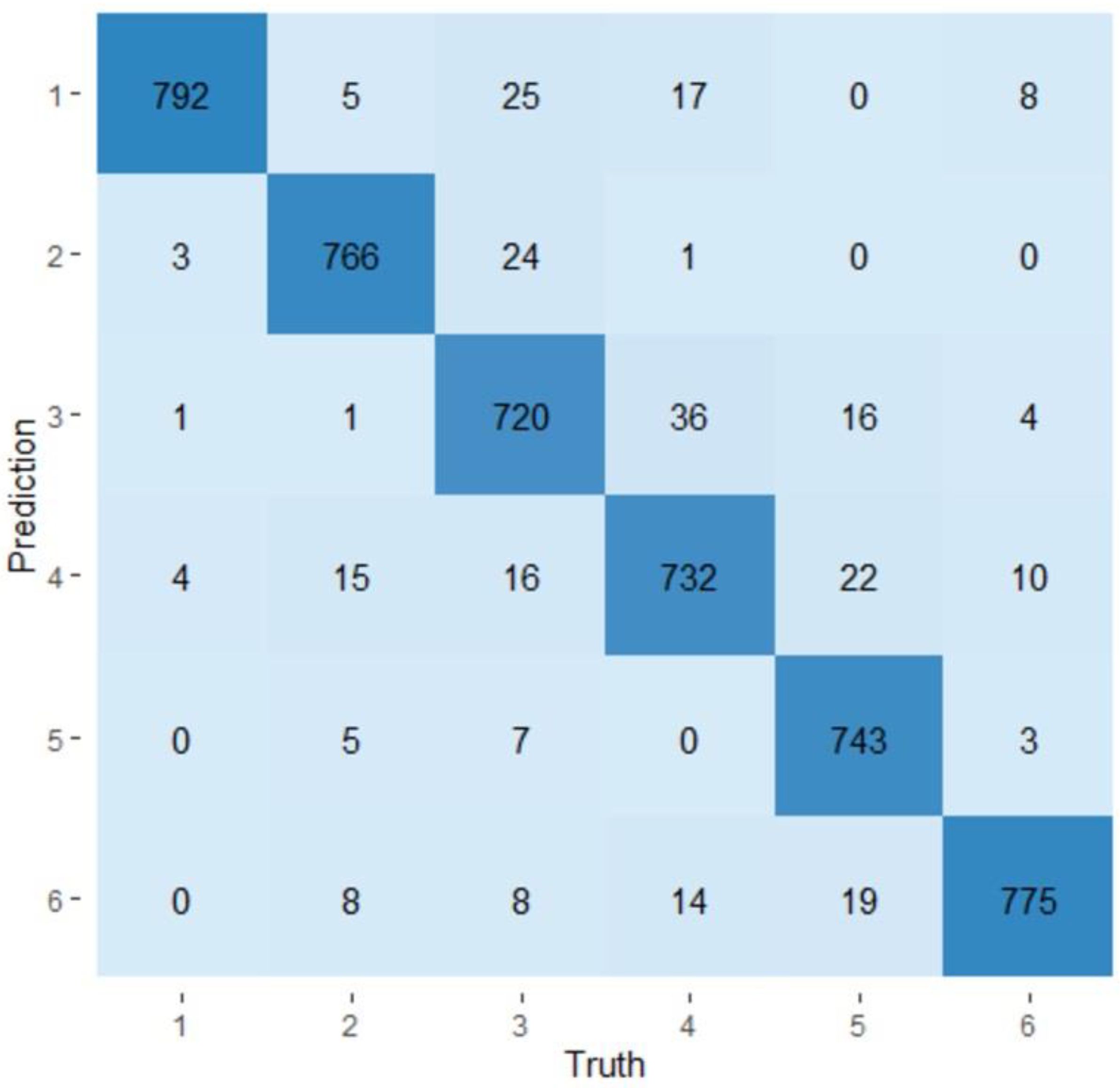
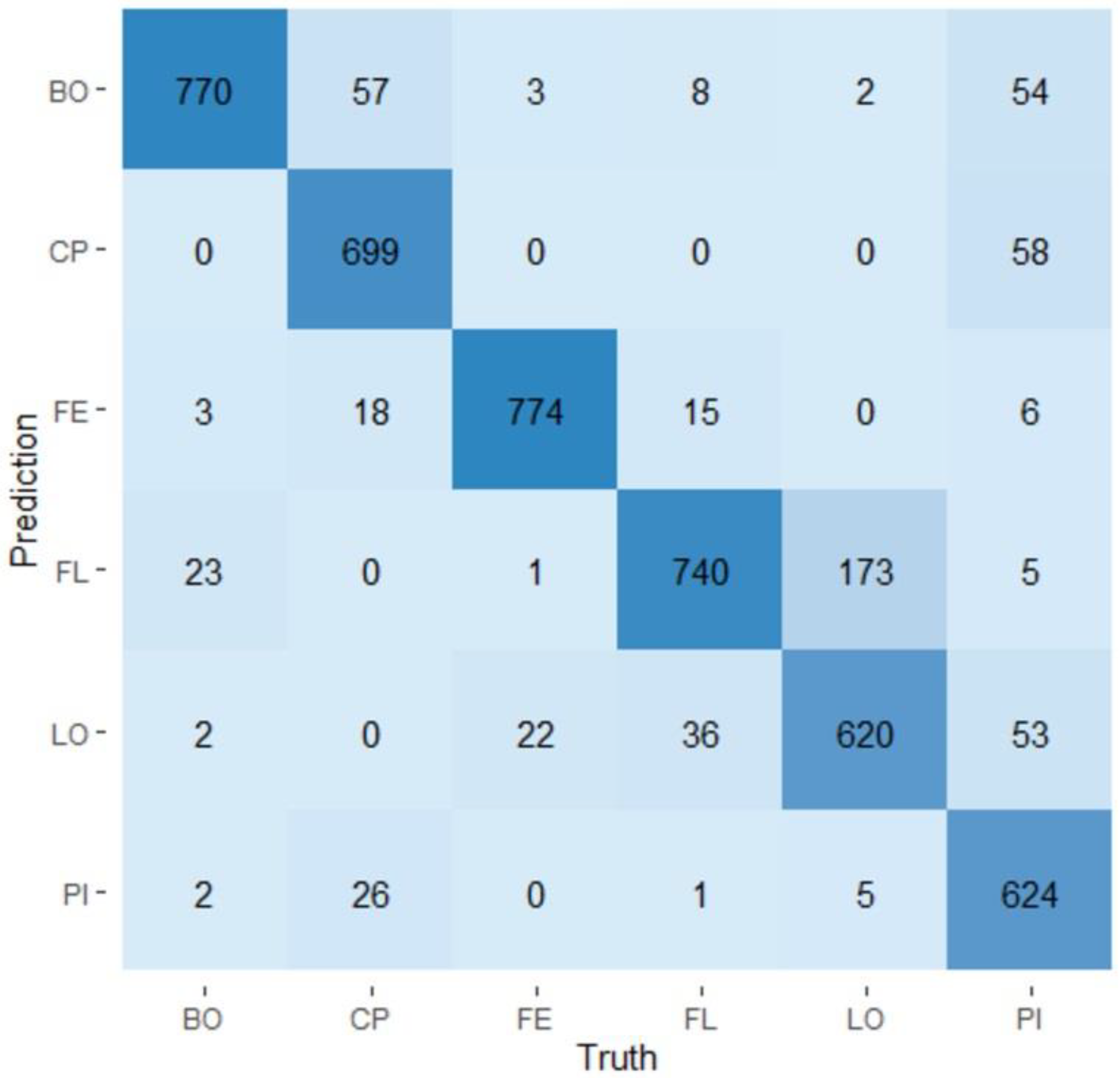
3.4. Random Forest
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bagge-Ravn, D. The microbial ecology of processing equipment in different fish industries—Analysis of the microflora during processing and following cleaning and disinfection. Int. J. Food Microbiol. 2003, 87, 239–250. [Google Scholar] [CrossRef]
- Public Health Agency of Canada. Food-Related Illnesses, Hospitalizations and Deaths in Canada; Government of Canada: Gatineau, QC, Canada, 2016.
- Environmental and Climate Change Canada. Taking Stock: Reducing Food Loss and Waste in Canada; Government of Canada: Gatineau, QC, Canada, 2019.
- Zwirzitz, B.; Wetzels, S.U.; Dixon, E.D.; Stessl, B.; Zaiser, A.; Rabanser, I.; Thalguter, S.; Pinior, B.; Roch, F.-F.; Strachan, C.; et al. The sources and transmission routes of microbial populations throughout a meat processing facility. NPJ Biofilms Microbiomes 2020, 6, 1–12. [Google Scholar] [CrossRef]
- Huis in’t Veld, J.H.J. Microbial and biochemical spoilage of foods: An overview. Int. J. Food Microbiol. 1996, 33, 1–18. [Google Scholar] [CrossRef]
- Havelaar, A.H.; Kirk, M.D.; Torgerson, P.R.; Gibb, H.J.; Hald, T.; Lake, R.J.; Praet, N.; Bellinger, D.C.; de Silva, N.R.; Gargouri, N.; et al. World Health Organization Global Estimates and Regional Comparisons of the Burden of Foodborne Disease in 2010. PLoS Med. 2015, 12, e1001923. [Google Scholar] [CrossRef] [Green Version]
- Zwirzitz, B.; Wetzels, S.U.; Dixon, E.D.; Fleischmann, S.; Selberherr, E.; Thalguter, S.; Quijada, N.M.; Dzieciol, M.; Wagner, M.; Stessl, B. Co-Occurrence of Listeria spp. and Spoilage Associated Microbiota During Meat Processing Due to Cross-Contamination Events. Front. Microbiol. 2021, 12, 632935. [Google Scholar] [CrossRef]
- Authority, E.F.S.J.E.J. The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2016. EFSA J. 2017, 15, e05077. [Google Scholar]
- Pothakos, V.; Devlieghere, F.; Villani, F.; Björkroth, J.; Ercolini, D. Lactic acid bacteria and their controversial role in fresh meat spoilage. Meat Sci. 2015, 109, 66–74. [Google Scholar] [CrossRef]
- Nychas, G.-J.E.; Skandamis, P.N.; Tassou, C.C.; Koutsoumanis, K.P. Meat spoilage during distribution. Meat Sci. 2008, 78, 77–89. [Google Scholar] [CrossRef]
- Verraes, C.; Van Boxstael, S.; Van Meervenne, E.; Van Coillie, E.; Butaye, P.; Catry, B.; De Schaetzen, M.A.; Van Huffel, X.; Imberechts, H.; Dierick, K.; et al. Antimicrobial resistance in the food chain: A review. Int. J. Environ. Res. Public Health 2013, 10, 2643–2669. [Google Scholar] [CrossRef] [Green Version]
- Møretrø, T.; Langsrud, S. Residential Bacteria on Surfaces in the Food Industry and Their Implications for Food Safety and Quality. Compr. Rev. Food Sci. Food Saf. 2017, 16, 1022–1041. [Google Scholar] [CrossRef] [Green Version]
- Giaouris, E.; Heir, E.; Desvaux, M.; Hébraud, M.; Møretrø, T.; Langsrud, S.; Doulgeraki, A.; Nychas, G.-J.; Kačániová, M.; Czaczyk, K.; et al. Intra- and inter-species interactions within biofilms of important foodborne bacterial pathogens. Front. Microbiol. 2015, 6, 841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stellato, G.; La Storia, A.; De Filippis, F.; Borriello, G.; Villani, F.; Ercolini, D. Overlap of Spoilage-Associated Microbiota between Meat and the Meat Processing Environment in Small-Scale and Large-Scale Retail Distributions. Appl. Environ. Microbiol. 2016, 82, 4045–4054. [Google Scholar] [CrossRef] [Green Version]
- Hultman, J.; Rahkila, R.; Ali, J.; Rousu, J.; Bjorkroth, J. Meat Processing Plant Microbiome and Contamination Patterns of Cold-Tolerant Bacteria Causing Food Safety and Spoilage Risks in the Manufacture of Vacuum-Packaged Cooked Sausages. Appl. Environ. Microbiol. 2015, 81, 7088–7097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.M.; Park, H.J.; Jang, H.I.; Kim, S.A.; Imm, J.Y.; Hwang, I.G.; Rhee, M.S. Changes in microbial contamination levels of porcine carcasses and fresh pork in slaughterhouses, processing lines, retail outlets, and local markets by commercial distribution. Res. Vet. Sci. 2013, 94, 413–418. [Google Scholar] [CrossRef]
- Stellato, G.; La Storia, A.; Cirillo, T.; Ercolini, D. Bacterial biogeographical patterns in a cooking center for hospital foodservice. Int. J. Food Microbiol. 2015, 193, 99–108. [Google Scholar] [CrossRef]
- Lim, E.S.; Kim, J.J.; Sul, W.J.; Kim, J.-S.; Kim, B.; Kim, H.; Koo, O.K. Metagenomic Analysis of Microbial Composition Revealed Cross-Contamination Pathway of Bacteria at a Foodservice Facility. Front. Microbiol. 2021, 12, 636329. [Google Scholar] [CrossRef]
- Smith, A.; Sterba-Boatwright, B.; Mott, J. Novel application of a statistical technique, Random Forests, in a bacterial source tracking study. Water Res. 2010, 44, 4067–4076. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Bergsveinson, J.; Ziola, B.; Mills, D.A. Mapping microbial ecosystems and spoilage-gene flow in breweries highlights patterns of contamina-tion and resistance. eLife 2015, 4, e04634. [Google Scholar] [CrossRef]
- Midelet, G.; Carpentier, B. Transfer of microorganisms, including Listeria monocytogenes, from various materials to beef. Appl. Environ. Microbiol. 2002, 68, 4015–4024. [Google Scholar] [CrossRef] [Green Version]
- Fagerlund, A.; Møretrø, T.; Heir, E.; Briandet, R.; Langsrud, S. Cleaning and Disinfection of Biofilms Composed of Listeria monocytogenes and Background Microbiota from Meat Processing Surfaces. Appl. Environ. Microbiol. 2017, 83, e01046-17. [Google Scholar] [CrossRef] [Green Version]
- Bridier, A.; Sanchez-Vizuete, P.; Guilbaud, M.; Piard, J.-C.; Naïtali, M.; Briandet, R. Biofilm-associated persistence of food-borne pathogens. Food Microbiol. 2015, 45, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Giaouris, E.; Heir, E.; Hébraud, M.; Chorianopoulos, N.; Langsrud, S.; Møretrø, T.; Habimana, O.; Desvaux, M.; Renier, S.; Nychas, G.-J. Attachment and biofilm formation by foodborne bacteria in meat processing environments: Causes, implications, role of bacterial interactions and control by alternative novel methods. Meat Sci. 2014, 97, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Tolker-Nielsen, T.; Molin, S. Spatial Organization of Microbial Biofilm Communities. Microb. Ecol. 2000, 40, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Botta, C.; Ferrocino, I.; Pessione, A.; Cocolin, L.; Rantsiou, K. Spatiotemporal Distribution of the Environmental Microbiota in Food Processing Plants as Impacted by Cleaning and Sanitizing Procedures: The Case of Slaughterhouses and Gaseous Ozone. Appl. Environ. Microbiol. 2020, 86, e01861-20. [Google Scholar] [CrossRef]
- De Filippis, F.; La Storia, A.; Villani, F.; Ercolini, D. Exploring the Sources of Bacterial Spoilers in Beefsteaks by Culture-Independent High-Throughput Se-quencing. PLoS ONE 2013, 8, e70222. [Google Scholar] [CrossRef] [Green Version]
- Shedleur-Bourguignon, F.; Thériault, W.P.; Longpré, J.; Thibodeau, A.; Fravalo, P. Use of an Ecosystem-Based Approach to Shed Light on the Heterogeneity of the Contamina-tion Pattern of Listeria monocytogenes on Conveyor Belt Surfaces in a Swine Slaughterhouse in the Province of Quebec, Canada. Pathogens 2021, 10, 1368. [Google Scholar] [CrossRef] [PubMed]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterio-plankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine mi-crobiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef]
- Larivière-Gauthier, G.; Thibodeau, A.; Letellier, A.; Yergeau, A.; Fravalo, P. Reduction of Salmonella Shedding by Sows during Gestation in Relation to Its Fecal Microbiome. Front. Microbiol. 2017, 8, 2219. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.; Wagner, H. Vegan: Community Ecology Package; R Package Version 2.5-6. 2018. Available online: https://CRAN.R-project.org/package=vegan (accessed on 28 December 2022).
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef] [PubMed]
- Duchemin, T.; Bar-Hen, A.; Lounissi, R.; Dab, W.; Hocine, M.N. Hierarchizing Determinants of Sick Leave: Insights From a Survey on Health and Well-being at the Workplace. J. Occup. Environ. Med. 2019, 61, e340–e347. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, M.; Vaughan, D. Yardstick: Tidy Characterizations of Model Performance. R package Version 0.0.8. 2021. Available online: https://CRAN.R-project.org/package=yardstick (accessed on 28 December 2022).
- Calero, G.C.; Gómez, N.C.; Lerma, L.L.; Benomar, N.; Knapp, C.W.; Abriouel, H. In silico mapping of microbial communities and stress responses in a porcine slaughterhouse and pork products through its production chain, and the efficacy of HLE disinfectant. Food Res. Int. 2020, 136, 109486. [Google Scholar] [CrossRef] [PubMed]
- Bridier, A.; Le Grandois, P.; Moreau, M.-H.; Prénom, C.; Le Roux, A.; Feurer, C.; Soumet, C. Impact of cleaning and disinfection procedures on microbial ecology and Salmonella antimicrobial re-sistance in a pig slaughterhouse. Sci. Rep. 2019, 9, 12947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Witte, C.; Flahou, B.; Ducatelle, R.; Smet, A.; De Bruyne, E.; Cnockaert, M.; Taminiau, B.; Daube, G.; Vandamme, P.; Haesebrouck, F. Detection, isolation and characterization of Fusobacterium gastrosuis sp. nov. colonizing the stomach of pigs. Syst. Appl. Microbiol. 2016, 40, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Wylensek, D.; Hitch, T.C.A.; Riedel, T.; Afrizal, A.; Kumar, N.; Wortmann, E.; Liu, T.; Devendran, S.; Lesker, T.R.; Hernández, S.B.; et al. A collection of bacterial isolates from the pig intestine reveals functional and taxonomic diversity. Nat. Commun. 2020, 11, 1–26. [Google Scholar] [CrossRef]
- Biasino, W.; De Zutter, L.; Mattheus, W.; Bertrand, S.; Uyttendaele, M.; Van Damme, I. Correlation between slaughter practices and the distribution of Salmonella and hygiene indicator bacteria on pig carcasses during slaughter. Food Microbiol. 2018, 70, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Stewart, P.S.; Hozalski, R.M. Biofilm Cohesive Strength as a Basis for Biofilm Recalcitrance: Are Bacterial Biofilms Overdesigned? Microbiol. Insights 2015, 8, S31444. [Google Scholar] [CrossRef]
- Braley, C.; Fravalo, P.; Gaucher, M.-L.; Larivière-Gauthier, G.; Shedleur-Bourguignon, F.; Longpré, J.; Thibodeau, A. Similar Carcass Surface Microbiota Observed Following Primary Processing of Different Pig Batches. Front. Microbiol. 2022, 13, 849883. [Google Scholar] [CrossRef]
- Hwang, B.K.; Choi, H.; Choi, S.H.; Kim, B.-S. Analysis of Microbiota Structure and Potential Functions Influencing Spoilage of Fresh Beef Meat. Front. Microbiol. 2020, 11, 1657. [Google Scholar] [CrossRef] [PubMed]
- Nychas, G.-J.; Marshall, D.L.; Sofos, J. Meat, Poultry, and Seafood. In Food Microbiology: Fundamentals and Frontiers; ASM Press: Washington, DC, USA, 2007; pp. 105–140. [Google Scholar]
- Remenant, B.; Jaffrès, E.; Dousset, X.; Pilet, M.-F.; Zagorec, M. Bacterial spoilers of food: Behavior, fitness and functional properties. Food Microbiol. 2015, 45, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Iulietto, M.F.; Sechi, P.; Borgogni, E.; Cenci-Goga, B.T. Meat Spoilage: A Critical Review of a Neglected Alteration Due to Ropy Slime Producing Bacteria. Ital. J. Anim. Sci. 2015, 14, 4011. [Google Scholar] [CrossRef]
- Doulgeraki, A.I.; Ercolini, D.; Villani, F.; Nychas, G.-J.E. Spoilage microbiota associated to the storage of raw meat in different conditions. Int. J. Food Microbiol. 2012, 157, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Betts, G. 23—Other Spoilage Bacteria, in Food Spoilage Microorganisms; Blackburn, C.d.W., Ed.; Woodhead Publishing: Sawston, UK, 2006; pp. 668–693. [Google Scholar]
- Moore, J.E.; Madden, R.H. Survival of Campylobacter coli in porcine liver. Food Microbiol. 2001, 18, 1–10. [Google Scholar] [CrossRef]
- Norton, D.M.; McCamey, M.A.; Gall, K.L.; Scarlett, J.M.; Boor, K.J.; Wiedmann, M. Molecular Studies on the Ecology of Listeria monocytogenes in the Smoked Fish Processing Industry. Appl. Environ. Microbiol. 2001, 67, 198–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laksanalamai, P.; Joseph, L.A.; Silk, B.J.; Burall, L.S.; LTarr, C.; Gerner-Smidt, P.; Datta, A.R. Genomic characterization of Listeria monocytogenes strains involved in a multistate listeriosis out-break associated with cantaloupe in US. PLoS ONE 2012, 7, e42448. [Google Scholar] [CrossRef]
- Barco, L.; Belluco, S.; Roccato, A.; Ricci, A. A systematic review of studies on Escherichia coli and Enterobacteriaceae on beef carcasses at the slaughter-house. Int. J. Food Microbiol. 2015, 207, 30–39. [Google Scholar] [CrossRef]
- Martínez-Avilés, M.; Garrido-Estepa, M.; Álvarez, J.; de la Torre, A. Salmonella Surveillance Systems in Swine and Humans in Spain: A Review. Veter Sci. 2019, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Self, J.L.; Luna-Gierke, R.E.; Fothergill, A.; Holt, K.G.; Vieira, A.R. Outbreaks attributed to pork in the United States, 1998–2015. Epidemiol. Infect. 2017, 145, 2980–2990. [Google Scholar] [CrossRef] [PubMed]
- Savariraj, W.R.; Ravindran, N.B.; Kannan, P.; Paramasivam, R.; Senthilkumar, T.M.A.; Kumarasamy, P.; Rao, V.A. Prevalence, antimicrobial susceptibility and virulence genes of Staphylococcus aureus isolated from pork meat in retail outlets in India. J. Food Saf. 2019, 39, e12589. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | V1 | V2 | V3 | V4 | V5 | V6 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % | Nbr | % | Nbr | % | Nbr | % | Nbr | % | Nbr | % | Nbr | |
| Fusobacterium | 16.66 | 42 | 10.47 | 24 | 14.39 | 39 | 12.72 | 21 | 12.97 | 34 | 13.94 | 34 |
| Trueperella | 11.27 | 23 | 13.17 | 20 | 13.62 | 26 | 11.98 | 17 | 13.50 | 27 | 13.82 | 31 |
| Pseudomonas | 10.31 | 17 | 12.45 | 23 | 11.1 | 21 | 13.49 | 32 | 8.88 | 24 | 10.15 | 20 |
| Acinetobacter | 14.32 | 22 | 11.56 | 20 | 12.51 | 22 | 12.22 | 34 | 8.80 | 20 | 12.19 | 20 |
| Peptoniphilus | 13.32 | 15 | 15.29 | 9 | 13.01 | 9 | 10.36 | 5 | 13.50 | 11 | 10.93 | 31 |
| Rothia | 11.19 | 13 | 12.8 | 17 | 10.12 | 5 | 7.43 | 5 | 8.56 | 6 | 9.36 | 5 |
| Enhydrobacter | 10.96 | 8 | 10.48 | 9 | 15.92 | 7 | ||||||
| Staphylococcus | 23.06 | 9 | 10.34 | 7 | ||||||||
| Bacteroides_unclassified | 12.12 | 24 | 9.88 | 11 | 7.54 | 11 | 12.53 | 5 | 11.45 | 26 | 9.79 | 12 |
| Aerococcaceae_unclassified | 6.77 | 6 | 12.45 | 12 | 12.58 | 16 | 13.00 | 12 | ||||
| Psychrobacter | 7.07 | 8 | 9.12 | 5 | 10.40 | 10 | ||||||
| Porphyromonas | 11.35 | 11 | 9.35 | 8 | 10.84 | 19 | 7.22 | 7 | 12.45 | 15 | 6.78 | 5 |
| Parvimonas | 5.9 | 5 | 7.57 | 17 | ||||||||
| Clostridium_sensu_stricto | 9.34 | 5 | 9.64 | 6 | 9.58 | 8 | ||||||
| Peptostreptococcus | 16.69 | 6 | ||||||||||
| Macrococcus | 8.98 | 8 | 18.58 | 9 | ||||||||
| Chryseobacterium | 6.74 | 6 | 7.25 | 9 | ||||||||
| Epilithonimonas | 11.6 | 7 | ||||||||||
| Prevotella | 7.43 | 10 | ||||||||||
| Clostridiaceae_1_unclassified | 6.64 | 5 | ||||||||||
| Genus | CP | FL | LO | BO | PI | FE | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % | Nbr | % | Nbr | % | Nbr | % | Nbr | % | Nbr | % | Nbr | |
| Fusobacterium | 14.63 | 28 | 24.46 | 33 | 17.24 | 21 | 16.32 | 44 | 23.48 | 35 | 16.36 | 33 |
| Trueperella | 15.39 | 19 | 24.28 | 36 | 11.35 | 6 | 12.17 | 41 | 14.51 | 27 | 10.49 | 15 |
| Pseudomonas | 9.17 | 32 | 7.68 | 10 | 8.03 | 22 | 9.24 | 22 | 9.34 | 13 | 17.04 | 38 |
| Acinetobacter | 10.57 | 32 | 10.34 | 13 | 21.39 | 22 | 14.75 | 24 | 12.3 | 17 | 13.5 | 30 |
| Peptoniphilus | 10.19 | 5 | 16.58 | 20 | 10.46 | 8 | 15.24 | 15 | 19.99 | 15 | 14.97 | 17 |
| Rothia | 10.27 | 28 | 34.35 | 7 | 10.98 | 11 | 6.92 | 5 | ||||
| Enhydrobacter | 14.52 | 9 | 15.74 | 5 | 14 | 5 | 10.82 | 5 | ||||
| Staphylococcus | 16.34 | 15 | ||||||||||
| Bacteroides_unclassified | 8.13 | 12 | 10.26 | 20 | 9.89 | 5 | 12.23 | 20 | 15.53 | 27 | 6.65 | 5 |
| Aerococcaceae_unclassified | 10.38 | 16 | 17.51 | 15 | 14.87 | 10 | 8.07 | 8 | ||||
| Psychrobacter | 10.23 | 16 | 5.73 | 8 | ||||||||
| Porphyromonas | 8.58 | 14 | 14.92 | 6 | 9.37 | 14 | 8.73 | 10 | 10.12 | 17 | ||
| Parvimonas | 7.06 | 6 | 7.93 | 6 | 6.43 | 11 | ||||||
| Clostridium_sensu_stricto | 10.46 | 18 | 9.26 | 5 | ||||||||
| Peptostreptococcus | 13.82 | 8 | ||||||||||
| Macrococcus | 11.91 | 24 | ||||||||||
| Streptococcus | 8.02 | 5 | ||||||||||
| Chryseobacterium | 7.87 | 9 | ||||||||||
| Epilithonimonas | 11.43 | 8 | ||||||||||
| Rhodopseudomonas | 5.98 | 9 | ||||||||||
| Prevotella | 7.94 | 6 | ||||||||||
| Clostridiaceae_1_unclassified | 6.62 | 5 | ||||||||||
| Observed | Shannon | Inv. Simpson | |
|---|---|---|---|
| V1 | 193 *V3, V4, V5, V6 | 2.97 *V2, V3, V4, V5 | 9.85 *V2, V4, V5 |
| V2 | 218 | 3.28 *V1, V6 | 13.64 *V1, V6 |
| V3 | 221 *V1 | 3.16 *V1, V4, V5 | 11.17 *V4, V5 |
| V4 | 239 *V1 | 3.39 *V1, V3, V6 | 14.51 *V1, V3, V6 |
| V5 | 245 *V1 | 3.47 *V1, V3, V6 | 16.5 *V1, V3, V6 |
| V6 | 226 *V1 | 3.07 *V2, V4, V5 | 10.35 *V2, V4, V5 |
| All visits p-values | 0.0838 | 0.0008 ** | 0.0003 ** |
| Observed | Shannon | Inv. Simpson | |
|---|---|---|---|
| CP | 372 *LO, FL, BO, PI, FE | 3.73 *LO, FL, BO, PI, FE | 18.5 *LO, FL, BO, PI, FE |
| LO | 169 *CP, PI, FE | 3.22 *CP, FL, BO | 11.87 *CP, FL |
| FL | 180 *CP, FE | 2.95 *CP, LO, FE | 9.18 *CP, LO, FE |
| BO | 167 *CP, PI, FE | 3.03 *CP, LO, FE | 10.46 *CP, FE |
| PI | 196 *CP, LO, BO, FE | 3.06 *CP, FE | 11.51 *CP |
| FE | 237 *CP, LO, FL, BO, PI | 3.29 *CP, FL, BO, PI | 13.52 *CP, FL, BO |
| All production lines p-values | 2.2 × 10−16 ** | 4.64 × 10−10 ** | 1.32 × 10−8 ** |
| Positive Associations | Negative Bacterial Associations | |||||
|---|---|---|---|---|---|---|
| Total | Unique | Relevant Bacterial Genera | Total | Unique | Relevant Bacterial Genera | |
| V1 | 0 | 0 | 0 | 0 | ||
| V2 | 14 | 4 | Listeria_80__Otu00067 | 20 | 1 | Escherichia_Shigella_Otu00046 |
| Brochothrix_Otu00245 | ||||||
| Streptococcus_Otu00029 | ||||||
| V3 | 39 | 10 | Acinetobacter_Otu00084 | 20 | 2 | Lactococcus_Otu00132 |
| Flavobacterium_Otu00354 | Streptococcus_Otu00029 | |||||
| Pseudomonas_Otu00173 | ||||||
| Psychrobacter_Otu00047 | ||||||
| V4 | 39 | 11 | Acinetobacter_Otu00084 | 23 | 1 | Escherichia_Shigella_Otu00046 |
| Enterococcus_77__Otu00071 | Aerococcus_Otu00053 | |||||
| Flavobacterium_Otu00138 | Lactococcus_Otu00132 | |||||
| Lactobacillus_Otu00092 | ||||||
| Psychrobacter_Otu00047 | ||||||
| Streptococcus_Otu00024 | ||||||
| V5 | 49 | 12 | Campylobacter_Otu00197 | 22 | 1 | Escherichia_Shigella_Otu00046 |
| Brochothrix_Otu00245 | Aerococcus_Otu00053 | |||||
| Enterococcus_77__Otu00071 | Lactococcus_Otu00132 | |||||
| Flavobacterium_Otu00354 | Streptococcus_Otu00029 | |||||
| Flavobacterium_Otu00138 | ||||||
| Flavobacterium_Otu00160 | ||||||
| Lactobacillus_Otu00092 | ||||||
| Pseudomonas_Otu00173 | ||||||
| Psychrobacter_Otu00047 | ||||||
| V6 | 33 | 8 | Campylobacter_Otu00197 | 24 | 4 | Escherichia_Shigella_Otu00046 |
| Campylobacter_Otu00233 | Brochothrix_Otu00245 | |||||
| Campylobacter_Otu00024 | Carnobacterium_58__Otu00055 | |||||
| Campylobacter_Otu00233 | Pseudomonas_Otu00037 | |||||
| Flavobacterium_Otu00160 | Streptococcus_Otu00029 | |||||
| Flavobacterium_Otu00354 | ||||||
| Flavobacterium_Otu00138 | ||||||
| Lactobacillus_Otu00092 | ||||||
| Psychrobacter_Otu00047 | ||||||
| Positive Associations | Negative Associations | |||||
|---|---|---|---|---|---|---|
| Total | Unique | Relevant Bacterial Genera | Total | Unique | Relevant Bacterial Genera | |
| CP | 169 | 97 | Campylobacter_Otu00493 | 13 | 2 | Acinetobacter_Otu00020 |
| Clostridium_sensu_stricto_Otu00016 | ||||||
| Clostridium_sensu_stricto_Otu00136 | ||||||
| Clostridium_sensu_stricto_Otu00307 | ||||||
| Clostridium_sensu_stricto_Otu00441 | ||||||
| Clostridium_sensu_stricto_Otu00059 | ||||||
| Clostridium_XlVa_75__Otu00385 | ||||||
| Enterococcus_77__Otu00071 | ||||||
| Flavobacterium_Otu00480 | ||||||
| Flavobacterium_Otu00333 | ||||||
| Flavobacterium_Otu00160 | ||||||
| Lactobacillus_Otu00092, Otu00099 | ||||||
| Lactococcus_Otu00262, Otu00132 | ||||||
| Moraxella_Otu00259 | ||||||
| Pseudomonas_Otu00179, Otu00003 | ||||||
| Psychrobacter_Otu00047 | ||||||
| Psychrobacter_Otu00012 | ||||||
| Psychrobacter_Otu00093 | ||||||
| FL | 42 | 14 | Clostridium_sensu_stricto_Otu00016 | 42 | 2 | Acinetobacter_Otu00011, Otu0020 |
| Pseudomonas_Otu00003, Otu00037 | ||||||
| Psychrobacter_Otu00012, Otu00047 | ||||||
| Moraxella_Otu00045 | ||||||
| LO | 36 | 11 | Clostridium_sensu_stricto_Otu00016 | 40 | 4 | Acinetobacter_Otu00020, Otu00011 |
| Enterococcus_77__Otu00071 | Moraxella_Otu00045 | |||||
| Flavobacterium_Otu00138 | ||||||
| Staphylococcus_99__Otu00008 | ||||||
| BO | 0 | 0 | 0 | 0 | ||
| PI | 29 | 5 | Clostridium_sensu_stricto_97__Otu00203 | 16 | 5 | Salmonella_66__Otu00026 |
| Psychrobacter_Otu00047 | Acinetobacter_Otu00020, Otu00011 | |||||
| Pseudomonas_Otu00023 | ||||||
| FE | 45 | 8 | Clostridium_sensu_stricto_Otu00059 | 21 | 0 | Moraxella_Otu00045 |
| Clostridium_sensu_stricto_Otu00016 | Morganella_91__Otu00337 | |||||
| Clostridium_sensu_stricto_Otu00136 | ||||||
| Campylobacter_Otu00493 | ||||||
| Escherichia_Shigella_Otu00046 | ||||||
| Carnobacterium_58__Otu00055 | ||||||
| Lactococcus_Otu00262 | ||||||
| Psychrobacter_Otu00012 | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shedleur-Bourguignon, F.; Duchemin, T.; P. Thériault, W.; Longpré, J.; Thibodeau, A.; Hocine, M.N.; Fravalo, P. Distinct Microbiotas Are Associated with Different Production Lines in the Cutting Room of a Swine Slaughterhouse. Microorganisms 2023, 11, 133. https://doi.org/10.3390/microorganisms11010133
Shedleur-Bourguignon F, Duchemin T, P. Thériault W, Longpré J, Thibodeau A, Hocine MN, Fravalo P. Distinct Microbiotas Are Associated with Different Production Lines in the Cutting Room of a Swine Slaughterhouse. Microorganisms. 2023; 11(1):133. https://doi.org/10.3390/microorganisms11010133
Chicago/Turabian StyleShedleur-Bourguignon, Fanie, Tom Duchemin, William P. Thériault, Jessie Longpré, Alexandre Thibodeau, Mounia N. Hocine, and Philippe Fravalo. 2023. "Distinct Microbiotas Are Associated with Different Production Lines in the Cutting Room of a Swine Slaughterhouse" Microorganisms 11, no. 1: 133. https://doi.org/10.3390/microorganisms11010133
APA StyleShedleur-Bourguignon, F., Duchemin, T., P. Thériault, W., Longpré, J., Thibodeau, A., Hocine, M. N., & Fravalo, P. (2023). Distinct Microbiotas Are Associated with Different Production Lines in the Cutting Room of a Swine Slaughterhouse. Microorganisms, 11(1), 133. https://doi.org/10.3390/microorganisms11010133