

Unveiling Rare Pathogens and Antibiotic Resistance in Tanzanian Cholera Outbreak Waters
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
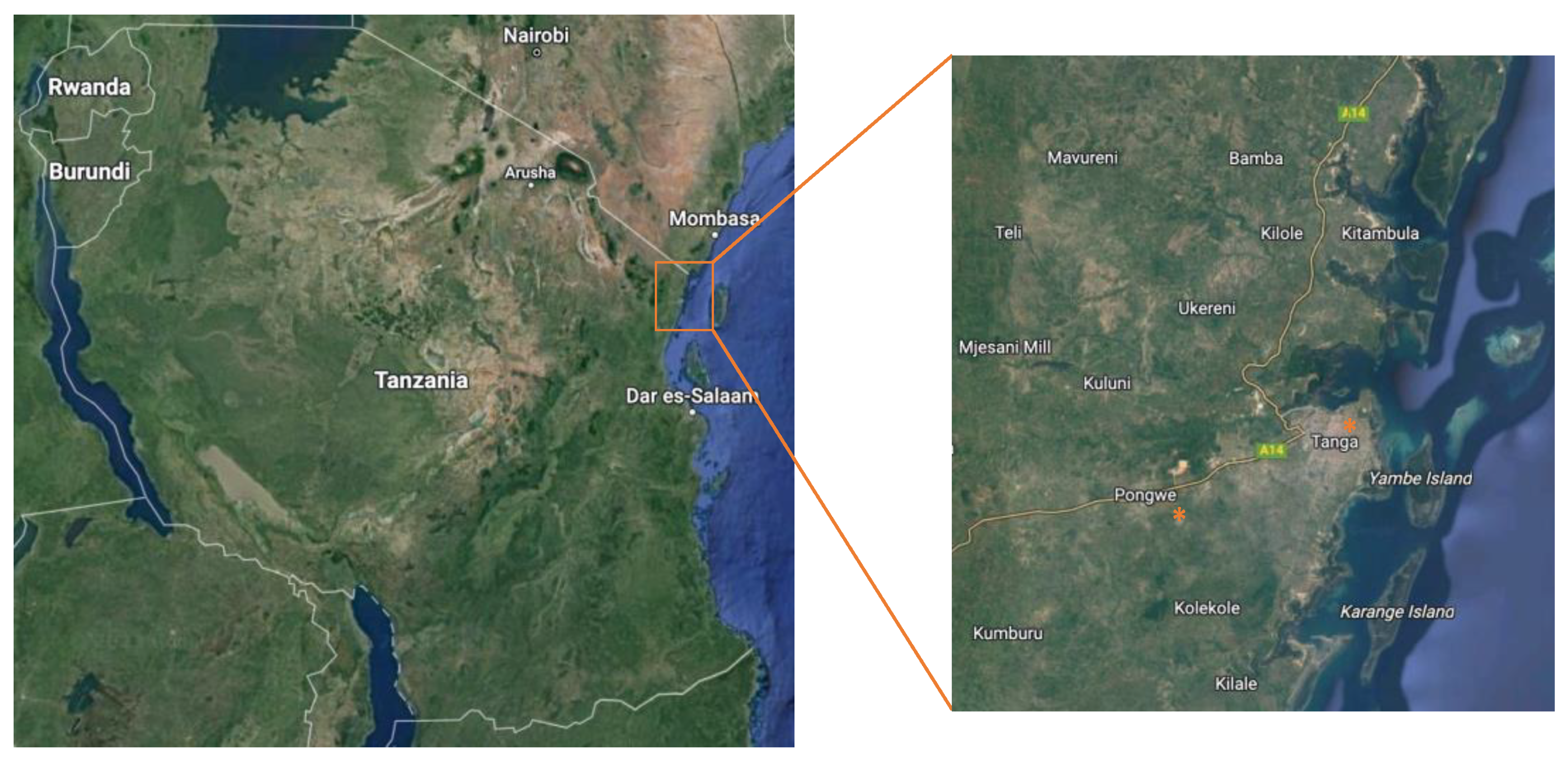
2.1. Collection of Environmental and Clean Water Samples
2.2. Ethical Considerations
2.3. Preparation of DNA for Bacteria and Bacteriophages
2.4. 16S/18S Amplicon-Based Microbiome Analysis
2.5. Oxford Nanopore Sequencing for Resistance Genes
2.6. Bioinformatics Analysis of Sequence Data
2.7. Statistics of Sequence Data
2.8. Digital Droplet PCR Amplification of Resistance Genes
3. Results
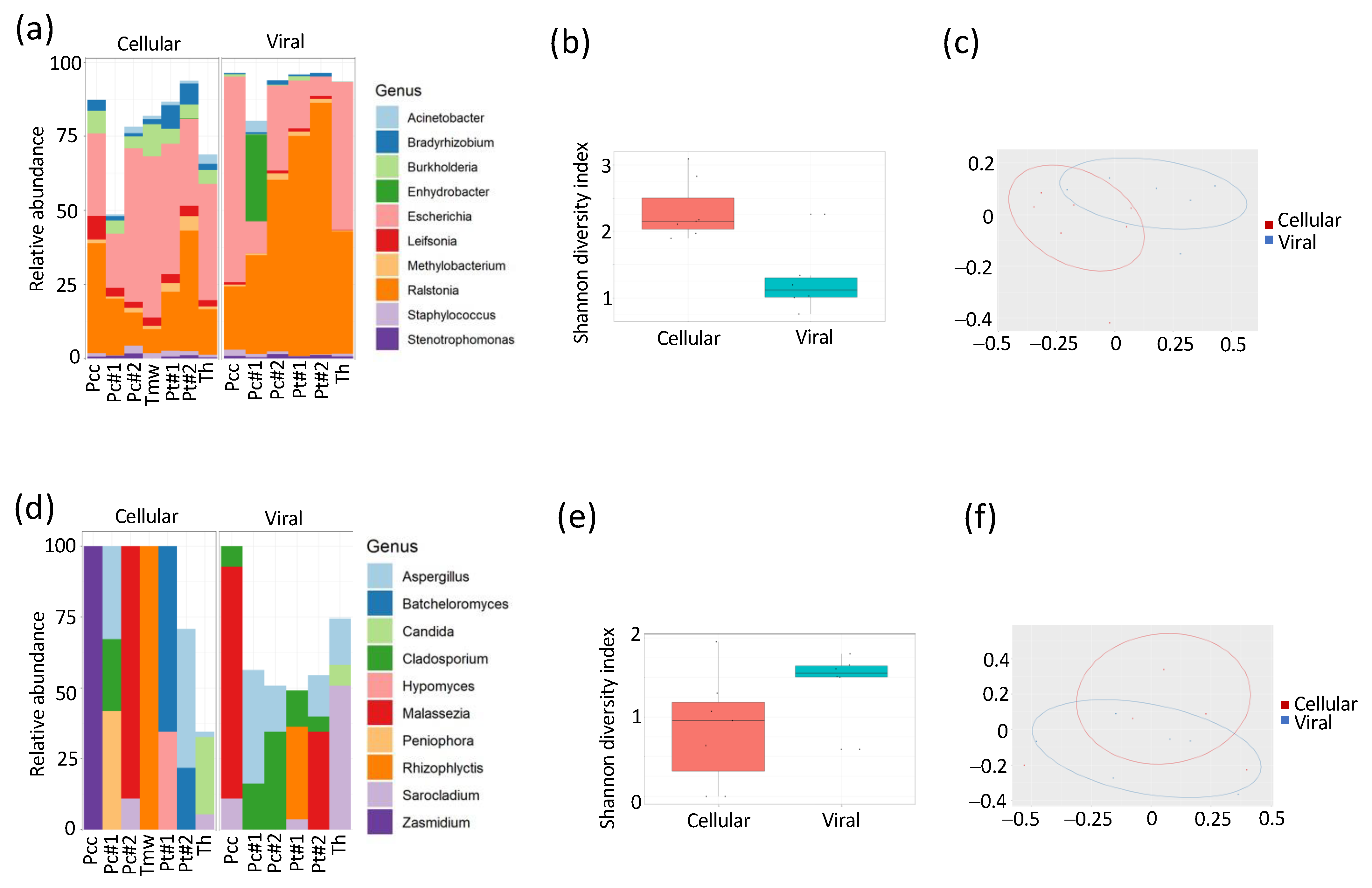
3.1. Composition of DNA Varies between Bacterial and Bacteriophage Compartments
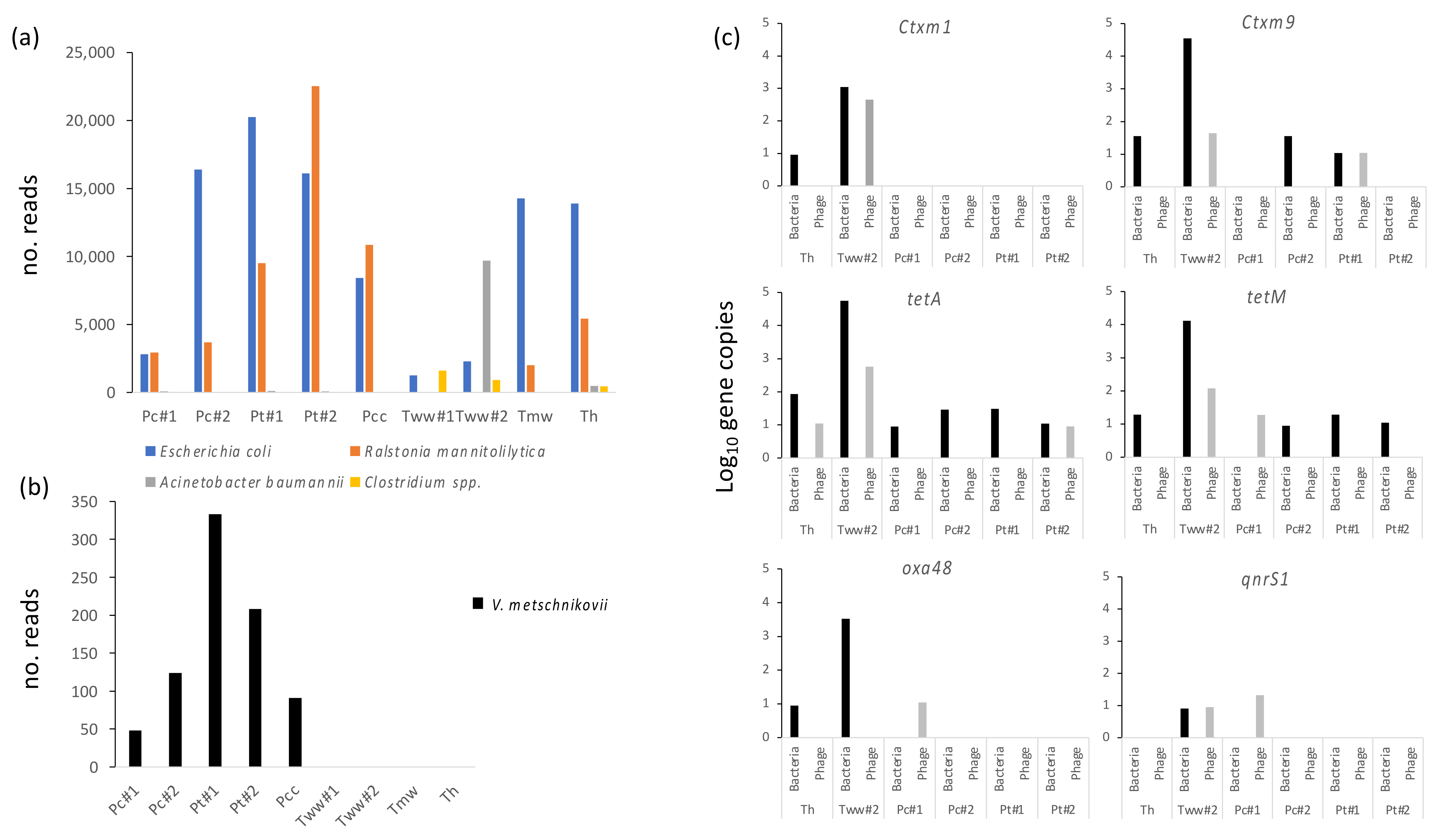
3.2. Water from Different Sources Is Heavily Contaminated with Classical Fecal, GI and Opportunistic Pathogens
3.3. Unusual Vibrios Are Likely Causative Agents of a Cholera Outbreak
3.4. Several Antibiotic Resistance Genes Are Identified in high Numbers in Both Bacterial and Bacteriophage Fractions from Water Sources
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dadgostar, P. Antimicrobial Resistance: Implications and Costs. Infect. Drug Resist. 2019, 12, 3903–3910. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Ministry of Agriculture, Livestock and Fisheries. National Action Plan for Antimicrobial Resistance 2017–2022; Ministry of Agriculture: Dar es Salaam, Tanzania, 2017; Volume 136.
- Ministry of Health. United Republic of Tanzania: The National Action Plan on Antimicrobial Resistance 2023–2028; Ministry of Health: Dodoma, Tanzania, 2022; p. 136.
- Velazquez-Meza, M.E.; Galarde-López, M.; Carrillo-Quiróz, B.; Alpuche-Aranda, C.M. Antimicrobial Resistance: One Health Approach. Vet. World 2022, 15, 743–749. [Google Scholar] [CrossRef] [PubMed]
- White, A.; Hughes, J.M. Critical Importance of a One Health Approach to Antimicrobial Resistance. Ecohealth 2019, 16, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Darby, E.M.; Trampari, E.; Siasat, P.; Gaya, M.S.; Alav, I.; Webber, M.A.; Blair, J.M.A. Molecular Mechanisms of Antibiotic Resistance Revisited. Nat. Rev. Microbiol. 2022, 21, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Vinayamohan, P.G.; Pellissery, A.J.; Venkitanarayanan, K. Role of Horizontal Gene Transfer in the Dissemination of Antimicrobial Resistance in Food Animal Production. Curr. Opin. Food Sci. 2022, 47, 100882. [Google Scholar] [CrossRef]
- Mikomangwa, W.P.; Bwire, G.M.; Kilonzi, M.; Mlyuka, H.; Mutagonda, R.F.; Kibanga, W.; Marealle, A.I.; Minzi, O.; Mwambete, K.D. The Existence of High Bacterial Resistance to Some Reserved Antibiotics in Tertiary Hospitals in Tanzania: A Call to Revisit Their Use. Infect. Drug Resist. 2020, 13, 1831–1838. [Google Scholar] [CrossRef]
- Seni, J.; Mapunjo, S.G.; Wittenauer, R.; Valimba, R.; Stergachis, A.; Werth, B.J.; Saitoti, S.; Mhadu, N.H.; Lusaya, E.; Konduri, N. Antimicrobial Use across Six Referral Hospitals in Tanzania: A Point Prevalence Survey. BMJ Open 2020, 10, e042819. [Google Scholar] [CrossRef]
- Mkinga, D.A.; Lyamuya, F.S. Aetiology and Antimicrobial Susceptibility Pattern of Bacteria Pathogens from Hospitalised Adult Patients at a Tertiary Care Hospital in North Eastern Tanzania. East Afr. Sci. 2022, 4, 54–61. [Google Scholar] [CrossRef]
- Sasi, P.; Mwakyandile, T.; Manyahi, J.; Kunambi, P.; Mugusi, S.; Rimoy, G. Ceftriaxone Prescribing and Resistance Pattern at a National Hospital in Tanzania. Tanzan. J. Health Res. 2020, 21, 1–13. [Google Scholar]
- Sasi, P.G. Ceftriaxone Prescription at Muhimbili National Hospital. Tanzan. J. Health Res. 2020, 21, 1–13. [Google Scholar] [CrossRef]
- World Bank. Tanzania Economic Update: Universal Access to Water and Sanitation Could Transform Social and Economic Development. Available online: https://www.worldbank.org/en/country/tanzania/publication/tanzania-economic-update-universal-access-to-water-and-sanitation-could-transform-social-and-economic-development (accessed on 25 June 2023).
- Chae, S.R.; Lukupulo, H.; Kim, S.; Walker, T.; Hardy, C.; Abade, A.; Urio, L.J.; Mghamba, J.; Quick, R. An Assessment of Household Knowledge and Practices during a Cholera Epidemic—Dar Es Salaam, Tanzania, 2016. Am. J. Trop. Med. Hyg. 2022, 107, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Olago, D.; Marshall, M.; Wandiga, S.O.; Opondo, M.; Yanda, P.Z.; Kangalawe, R.; Githeko, A.; Downs, T.; Opere, A.; Kabumbuli, R.; et al. Climatic, Socio-Economic, and Health Factors Affecting Human Vulnerability to Cholera in the Lake Victoria Basin, East Africa. AMBIO 2007, 36, 350–358. [Google Scholar] [CrossRef]
- Asadgol, Z.; Mohammadi, H.; Kermani, M.; Badirzadeh, A.; Gholami, M. The Effect of Climate Change on Cholera Disease: The Road Ahead Using Artificial Neural Network. PLoS ONE 2019, 14, e0224813. [Google Scholar] [CrossRef]
- MacFadden, D.R.; McGough, S.F.; Fisman, D.; Santillana, M.; Brownstein, J.S. Antibiotic Resistance Increases with Local Temperature. Nat. Clim. Chang. 2018, 8, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Karkman, A.; Do, T.T.; Walsh, F.; Virta, M.P.J. Antibiotic-Resistance Genes in Waste Water. Trends Microbiol. 2018, 26, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Muniesa, M.; Colomer-Lluch, M.; Jofre, J. Potential Impact of Environmental Bacteriophages in Spreading Antibiotic Resistance Genes. Future Microbiol. 2013, 8, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Hughes, D. Antibiotic Resistance and Its Cost: Is It Possible to Reverse Resistance? Nat. Rev. Microbiol. 2010, 8, 260–271. [Google Scholar] [CrossRef]
- Torres-Barceló, C. The Disparate Effects of Bacteriophages on Antibiotic-Resistant Bacteria. Emerg. Microbes Infect. 2018, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lood, R.; Ertürk, G.; Mattiasson, B. Revisiting Antibiotic Resistance Spreading in Wastewater Treatment Plants—Bacteriophages as a Much Neglected Potential Transmission Vehicle. Front. Microbiol. 2017, 8, 2298. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Wang, B.; Zhang, X.; Zhang, J.; Zhang, H.; Liu, X.; Gao, Z.; Yu, Z. The Spread of Antibiotic Resistance to Humans and Potential Protection Strategies. Ecotoxicol. Environ. Saf. 2023, 254, 114734. [Google Scholar] [CrossRef] [PubMed]
- Quirós, P.; Colomer-Lluch, M.; Martínez-Castillo, A.; Miró, E.; Argente, M.; Jofre, J.; Navarro, F.; Muniesa, M. Antibiotic Resistance Genes in the Bacteriophage DNA Fraction of Human Fecal Samples. Antimicrob. Agents Chemother. 2014, 58, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Karkman, A.; Pärnänen, K.; Larsson, D.G.J. Fecal Pollution Can Explain Antibiotic Resistance Gene Abundances in Anthropogenically Impacted Environments. Nat. Commun. 2019, 10, 80. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Zhang, L. Wastewater Pathogen Surveillance Based on One Health Approach. Lancet Microbe 2023, 4, e297. [Google Scholar] [CrossRef]
- Kumburu, H.H.; Sonda, T.; Van Zwetselaar, M.; Leekitcharoenphon, P.; Lukjancenko, O.; Mmbaga, B.T.; Alifrangis, M.; Lund, O.; Aarestrup, F.M.; Kibiki, G.S. Using WGS to Identify Antibiotic Resistance Genes and Predict Antimicrobial Resistance Phenotypes in MDR Acinetobacter Baumannii in Tanzania. J. Antimicrob. Chemother. 2019, 74, 1484–1493. [Google Scholar] [CrossRef]
- Yadav, S.; Kapley, A. Antibiotic Resistance: Global Health Crisis and Metagenomics. Biotechnol. Rep. 2021, 29, e00604. [Google Scholar] [CrossRef] [PubMed]
- Owusu, M.; Acheampong, G.; Annan, A.; Marfo, K.S.; Osei, I.; Amuasi, J.; Sarpong, N.; Im, J.; Mogeni, O.D.; Chiang, H.Y.; et al. Ralstonia Mannitolilytica Sepsis: A Case Report. J. Med. Case Rep. 2019, 13, 318. [Google Scholar] [CrossRef]
- Agensi, A.; Tibyangye, J.; Tamale, A.; Agwu, E.; Amongi, C. Contamination Potentials of Household Water Handling and Storage Practices in Kirundo Subcounty, Kisoro District, Uganda. J. Environ. Public Health 2019, 2019, 7932193. [Google Scholar] [CrossRef]
- Berihun, G.; Abebe, M.; Hassen, S.; Gizeyatu, A.; Berhanu, L.; Teshome, D.; Walle, Z.; Desye, B.; Sewunet, B.; Keleb, A. Drinking Water Contamination Potential and Associated Factors among Households with Under-Five Children in Rural Areas of Dessie Zuria District, Northeast Ethiopia. Front. Public Health 2023, 11, 1199314. [Google Scholar] [CrossRef]
- Andersson, T.; Adell, A.D.; Moreno-Switt, A.A.; Spégel, P.; Turner, C.; Overballe-Petersen, S.; Fuursted, K.; Lood, R. Biogeographic variation in antimicrobial resistance in rivers is influenced by agriculture and is spread through bacteriophages. Environ. Microbiol. 2022, 24, 4869–4884. [Google Scholar] [CrossRef] [PubMed]
- Andersson, T.; Makenga, G.; Francis, F.; Minja, D.T.R.; Overballe-Petersen, S.; Tang, M.H.E.; Fuursted, K.; Baraka, V.; Lood, R. Enrichment of Antibiotic Resistance Genes within Bacteriophage Populations in Saliva Samples from Individuals Undergoing Oral Antibiotic Treatments. Front. Microbiol. 2022, 13, 1049110. [Google Scholar] [CrossRef]
- Ring, H.C.; Thorsen, J.; Saunte, D.M.; Lilje, B.; Bay, L.; Riis, P.T.; Larsen, N.; Andersen, L.O.; Nielsen, H.V.; Miller, I.M.; et al. The Follicular Skin Microbiome in Patients With Hidradenitis Suppurativa and Healthy Controls. JAMA Dermatol. 2017, 153, 897–905. [Google Scholar] [CrossRef]
- Loyola, S.; Loyola, S.; Sanchez, J.F.; Sanchez, J.F.; Maguiña, E.; Canal, E.; Castillo, R.; Bernal, M.; Meza, Y.; Tilley, D.H.; et al. Fecal Contamination of Drinking Water Was Associated with Diarrheal Pathogen Carriage among Children Younger than 5 Years in Three Peruvian Rural Communities. Am. J. Trop. Med. Hyg. 2020, 102, 1279. [Google Scholar] [CrossRef] [PubMed]
- Pickering, A.J.; Davis, J.; Walters, S.P.; Horak, H.M.; Keymer, D.P.; Mushi, D.; Strickfaden, R.; Chynoweth, J.S.; Liu, J.; Blum, A.; et al. Hands, Water, and Health: Fecal Contamination in Tanzanian Communities with Improved, Non-Networked Water Supplies. Environ. Sci. Technol. 2010, 44, 3267–3272. [Google Scholar] [CrossRef] [PubMed]
- Konechnyi, Y.; Khorkavyi, Y.; Ivanchuk, K.; Kobza, I.; Sękowska, A.; Korniychuk, O. Vibrio Metschnikovii: Current State of Knowledge and Discussion of Recently Identified Clinical Case. Clin. Case Rep. 2021, 9, 2236–2244. [Google Scholar] [CrossRef]
- Linde, H.J.; Kobuch, R.; Jayasinghe, S.; Reischl, U.; Lehn, N.; Kaulfuss, S.; Beutin, L. Vibrio Metschnikovii, a Rare Cause of Wound Infection. J. Clin. Microbiol. 2004, 42, 4909–4911. [Google Scholar] [CrossRef] [PubMed]
- Valáriková, J.; Korcová, J.; Ziburová, J.; Rosinský, J.; Čížová, A.; Bieliková, S.; Sojka, M.; Farkaš, P. Potential Pathogenicity and Antibiotic Resistance of Aquatic Vibrio Isolates from Freshwater in Slovakia. Folia Microbiol. 2019, 65, 545–555. [Google Scholar] [CrossRef]
- Magalhães, V.; Branco, A.; De Andrade Lima, R.; Magalhães, M. Vibrio Metschnikovii among Diarrheal Patients during Cholera Epidemic in Recife Brazil. Rev. Inst. Med. Trop. Sao Paulo 1996, 38, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Mohd Ali, M.R.; Zamri, H.F.; Nor Amdan, N.A.; Azmai, M.N.A.; Maniam, S.; Mohamed Alipiah, N.; Hashim, R. Distribution, Prevalence, and Antibiotic Susceptibility Profiles of Infectious Noncholera Vibrio Species in Malaysia. J. Trop. Med. 2023, 2023, 2716789. [Google Scholar] [CrossRef] [PubMed]
- Sonola, V.S.; Katakweba, A.; Misinzo, G.; Matee, M.I. Molecular Epidemiology of Antibiotic Resistance Genes and Virulence Factors in Multidrug-Resistant Escherichia Coli Isolated from Rodents, Humans, Chicken, and Household Soils in Karatu, Northern Tanzania. Int. J. Environ. Res. Public Health 2022, 19, 5388. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, E.; Bonnin, R.A.; Rocha, E.P.C. Phage-Plasmids Spread Antibiotic Resistance Genes through Infection and Lysogenic Conversion. mBio 2022, 13, e0185122. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Sample | District | Village | GPS Location |
|---|---|---|---|
| Pcc | Pongwe | Manzara | 5°08′38.5” S 38°58′29.2” E |
| Pt#1 | Pongwe | Manzara | 5°08′26.1” S 38°58′35.7” E |
| Pt#2 | Pongwe | Manzara | 5°08′26.1” S 38°58′35.7” E |
| Pc#1 | Pongwe | Manzara | 5°07′29.2” S 38°58′38.2” E |
| Pc#2 | Pongwe | Manzara | 5°07′29.2” S 38°58′38.2” E |
| Th | Tanga | Tanga | 5°03′47” N 39°06′44” E |
| Tmw | Tanga | Tanga | 5°03′47” N 39°06′44” E |
| Tww#1 | Tanga | Tanga | 5°03′47” N 39°06′44” E |
| Tww#2 | Tanga | Tanga | 5°03′47” N 39°06′44” E |
| Target | Forward Primer | Reverse Primer | Probe | Refs |
|---|---|---|---|---|
| 16S | AGAGTTTGATCCTGGCTCAGGA | CGTGTTACTCACCCGTCCG | CGCTGGCGGCGTGCCTAATACATGC | [33] |
| CTXM1 | ACAGTACAGCGATAACGTGG | GAATGGCGGTGTTTAACGTC | GCGGCCCGGCTAGCGTCACC | [34] |
| CTXM9 | GACTGTGGGTGATAAGACCG | TGTTGCGGCTGGGTAAAATA | GCAGGGTCGTGCGCCGCTGG | [34] |
| OXA48 | AAGTTACACGTATCGGAGCG | ACCAGCCAATCTTAGGTTCG | AGCCATGCTGACCGAAGCCAATGGTGA | [34] |
| tetA | TTGAACGGCCTCAATTTCCT | GATGAAGAAGACCGCCATCA | GCATGACCGTCGTCGCCGCCC | [33,34] |
| tetM | TGCAAGAAAAGTATCATGTGGAG | AAACCGAGCTCTCATACTGC | TGCCGCCAAATCCTTTCTGGGCTTCCA | [33,34] |
| qnrS1 | GGGTGCATCACTGAAAGAGT | CCAGTGCTTCGAGAATCAGT | TGCCACGCCGAACTCGACGGTTTAGA | [34] |
| 341F/806R | ACTCCTAYGGGRBGCASCAG | AGCGTGGACTACNNGGGTATCTAAT | [35] | |
| G3 (V3-V4) | GCCAGCAGCCGCGGTAATTC | ACATTCTTGGCAAATGCTTTCGCAG | [35] | |
| G4 (V3-V4) | CAGCCGCGGTAATTCCAGCTC | GGTGGTGCCCTTCCGTCAAT | [35] | |
| G6 (V3-V5) | TGGAGGGCAAGTCTGGTGCC | ACGGTATCTGATCGTCTTCGATCCC | [35] |
| Sample | Sample Source | Antibiotic Group | Resistance Gene |
|---|---|---|---|
| Tww1 | Tetracycline | tet(C, M, Q, W) | |
| Beta-lactam | blaTEM-1A, blaOXA, cfx(A3, A46) | ||
| Tanga, Wastewater | Macrolide | erm(G), msr(D), med(A) | |
| Aminoglycoside | aac(6′), aph(2″), aph(3′)-Ia | ||
| Phenicol | catQ | ||
| Pcc | Pongwe, well | Tetracycline | tet(C, M) |
| Beta-lactam | blaTEM-1A, blaOXA-22 | ||
| Tmw | Tetracycline | tet(C, K) | |
| Beta-lactam | blaTEM-1A, blaOXA-22 | ||
| Tanga, tap | |||
| Medical ward | Aminoglycoside | Aph(3′)-1a | |
| Phenicol | catA1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baraka, V.; Andersson, T.; Makenga, G.; Francis, F.; Minja, D.T.R.; Overballe-Petersen, S.; Tang, M.-H.E.; Fuursted, K.; Lood, R. Unveiling Rare Pathogens and Antibiotic Resistance in Tanzanian Cholera Outbreak Waters. Microorganisms 2023, 11, 2490. https://doi.org/10.3390/microorganisms11102490
Baraka V, Andersson T, Makenga G, Francis F, Minja DTR, Overballe-Petersen S, Tang M-HE, Fuursted K, Lood R. Unveiling Rare Pathogens and Antibiotic Resistance in Tanzanian Cholera Outbreak Waters. Microorganisms. 2023; 11(10):2490. https://doi.org/10.3390/microorganisms11102490
Chicago/Turabian StyleBaraka, Vito, Tilde Andersson, Geofrey Makenga, Filbert Francis, Daniel T. R. Minja, Sören Overballe-Petersen, Man-Hung Eric Tang, Kurt Fuursted, and Rolf Lood. 2023. "Unveiling Rare Pathogens and Antibiotic Resistance in Tanzanian Cholera Outbreak Waters" Microorganisms 11, no. 10: 2490. https://doi.org/10.3390/microorganisms11102490