

Clostridioides difficile from Fecally Contaminated Environmental Sources: Resistance and Genetic Relatedness from a Molecular Epidemiological Perspective

Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Identification of C. difficile
2.2. PCR-Ribotyping and Toxin Genotyping
2.3. Antimicrobial Susceptibility Testing
2.4. Whole Genome Sequencing and Data Analysis
3. Results
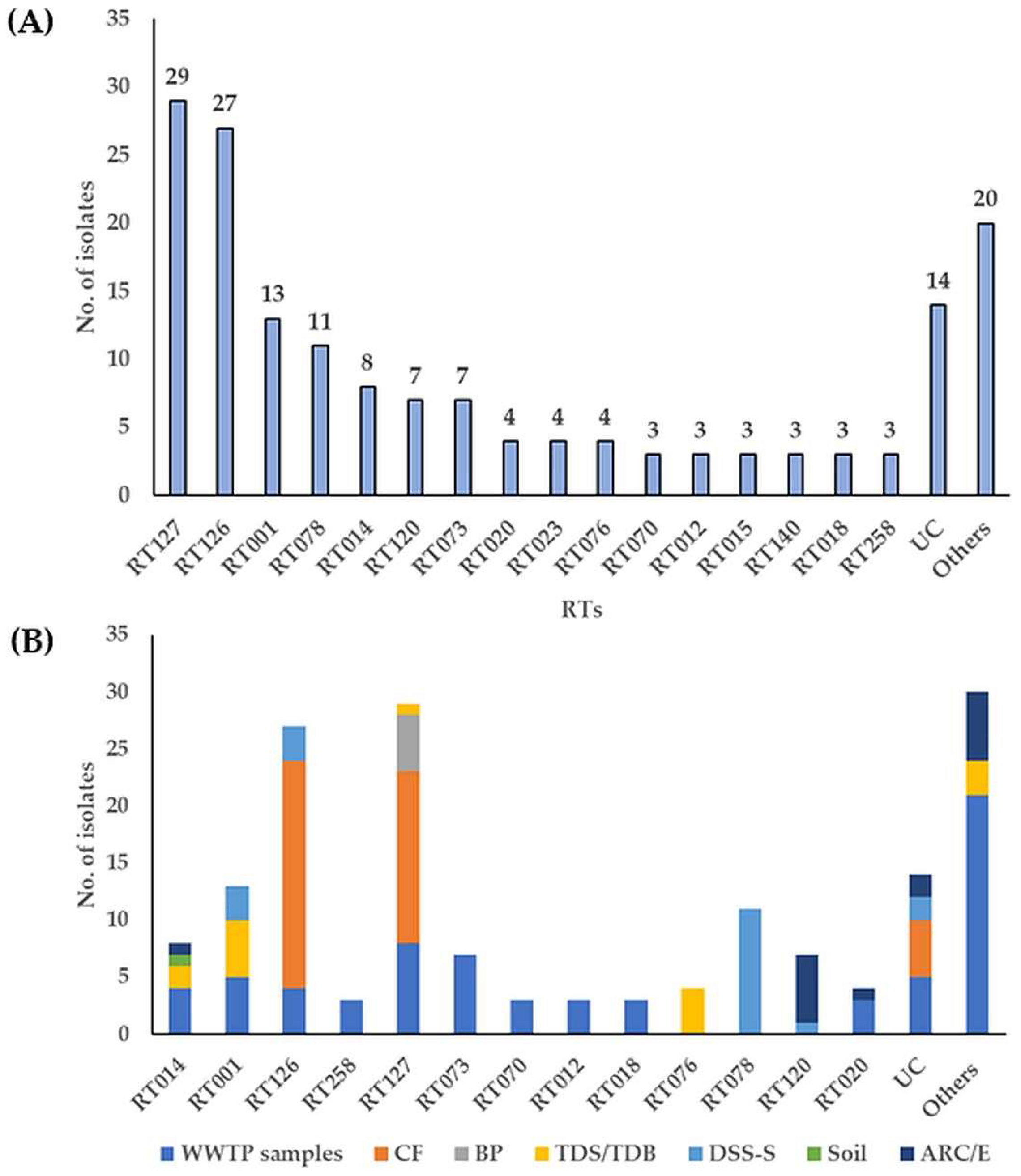
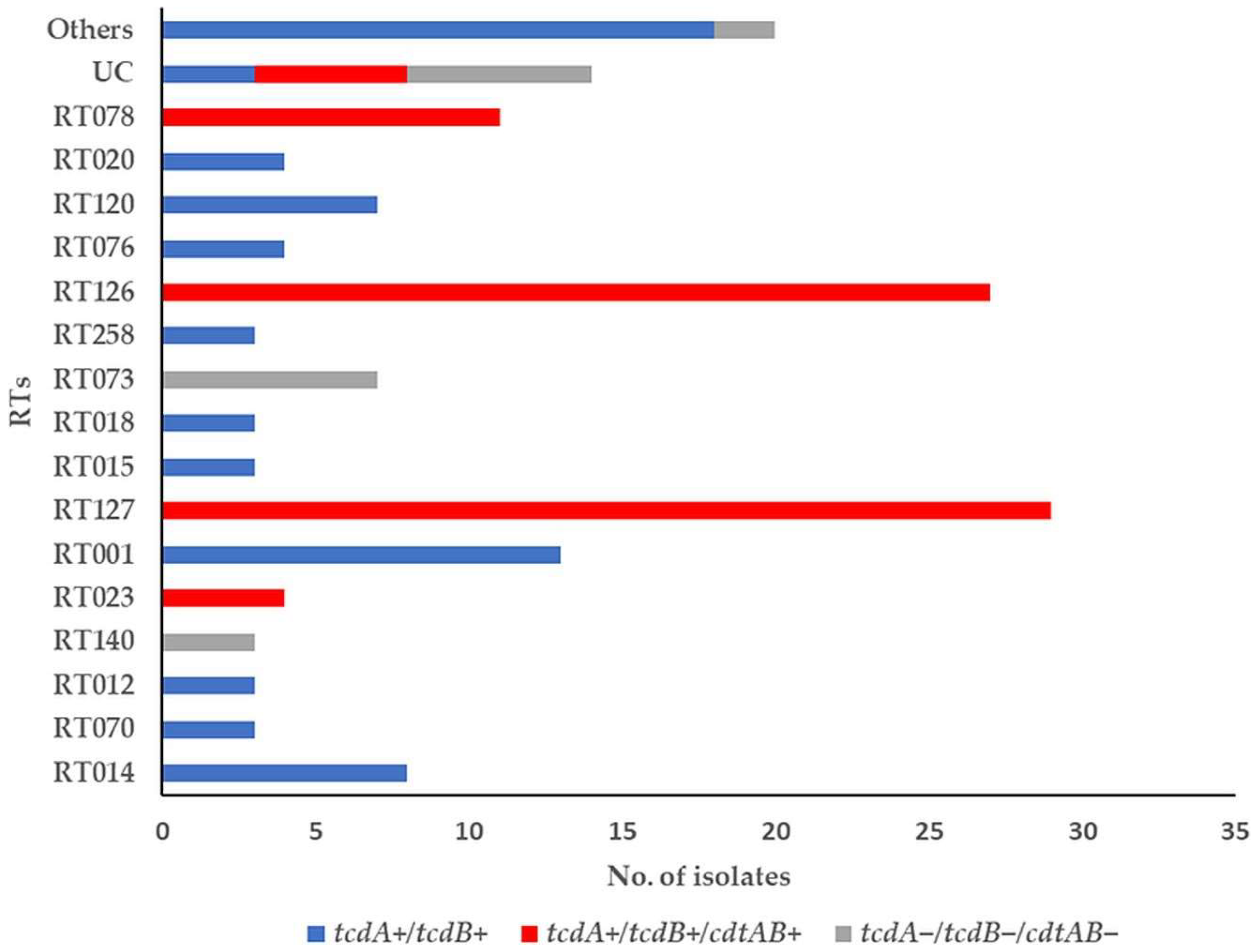
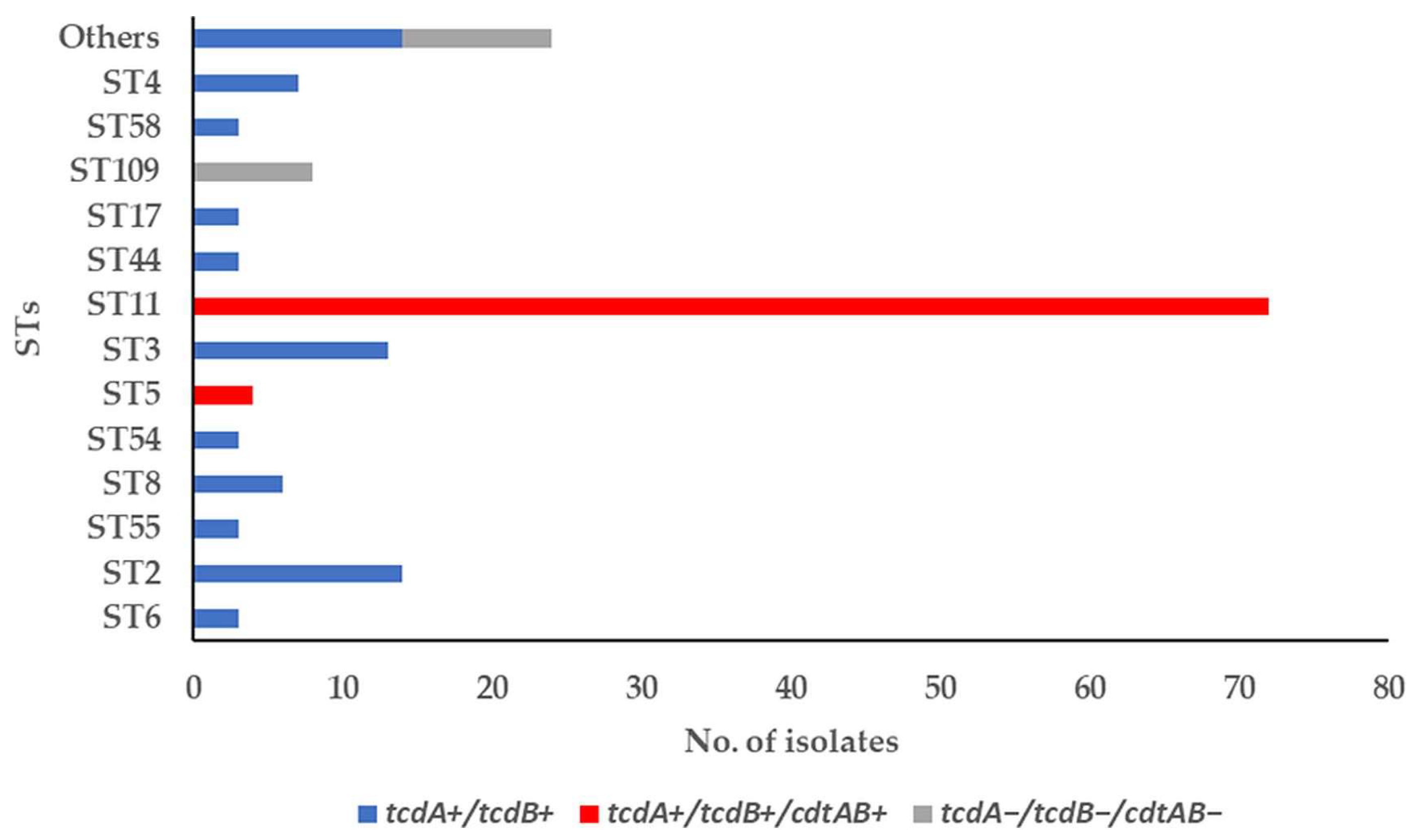
3.1. Toxin-Encoding Genes and PCR Ribotypes of C. difficile Strains
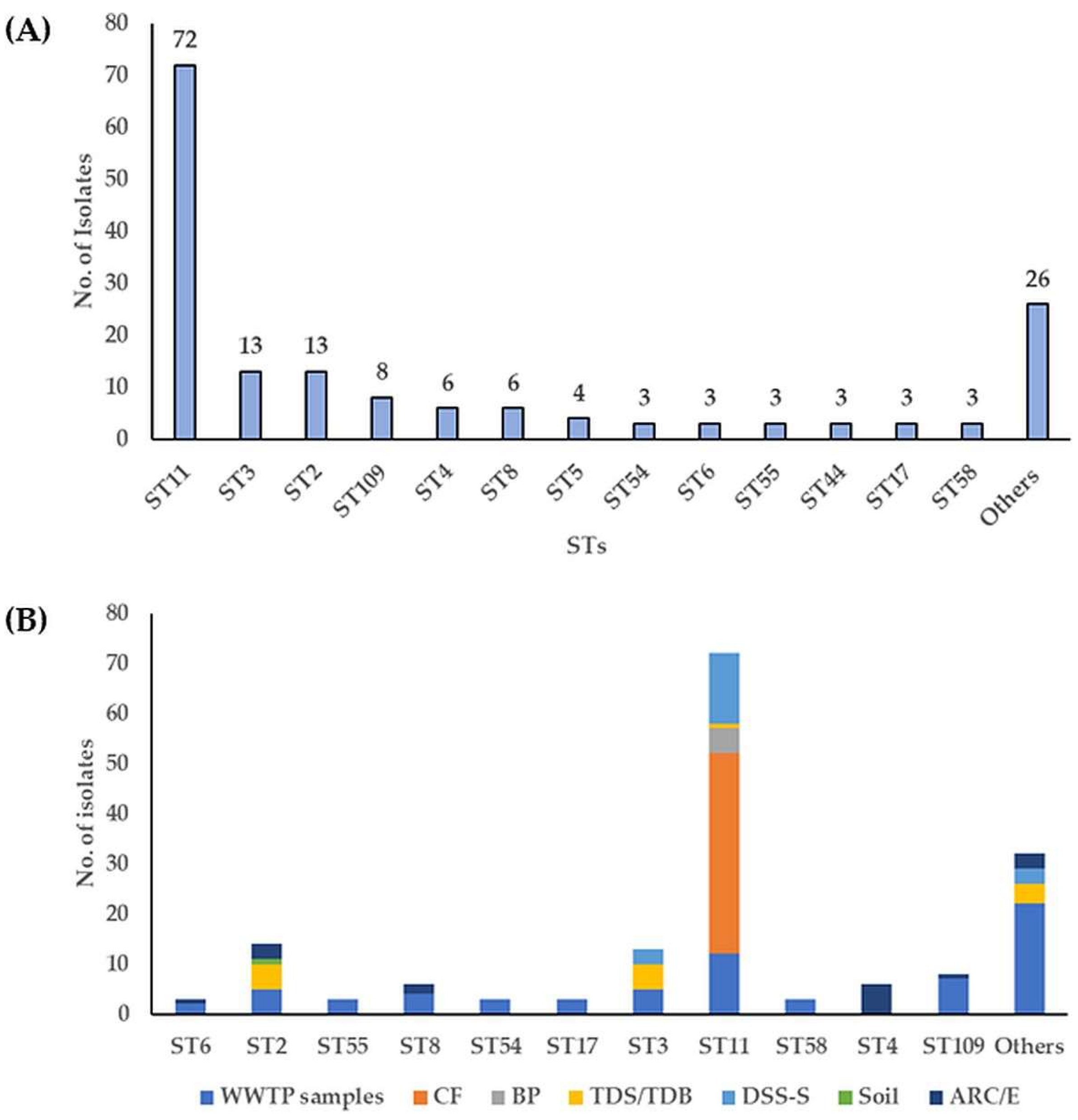
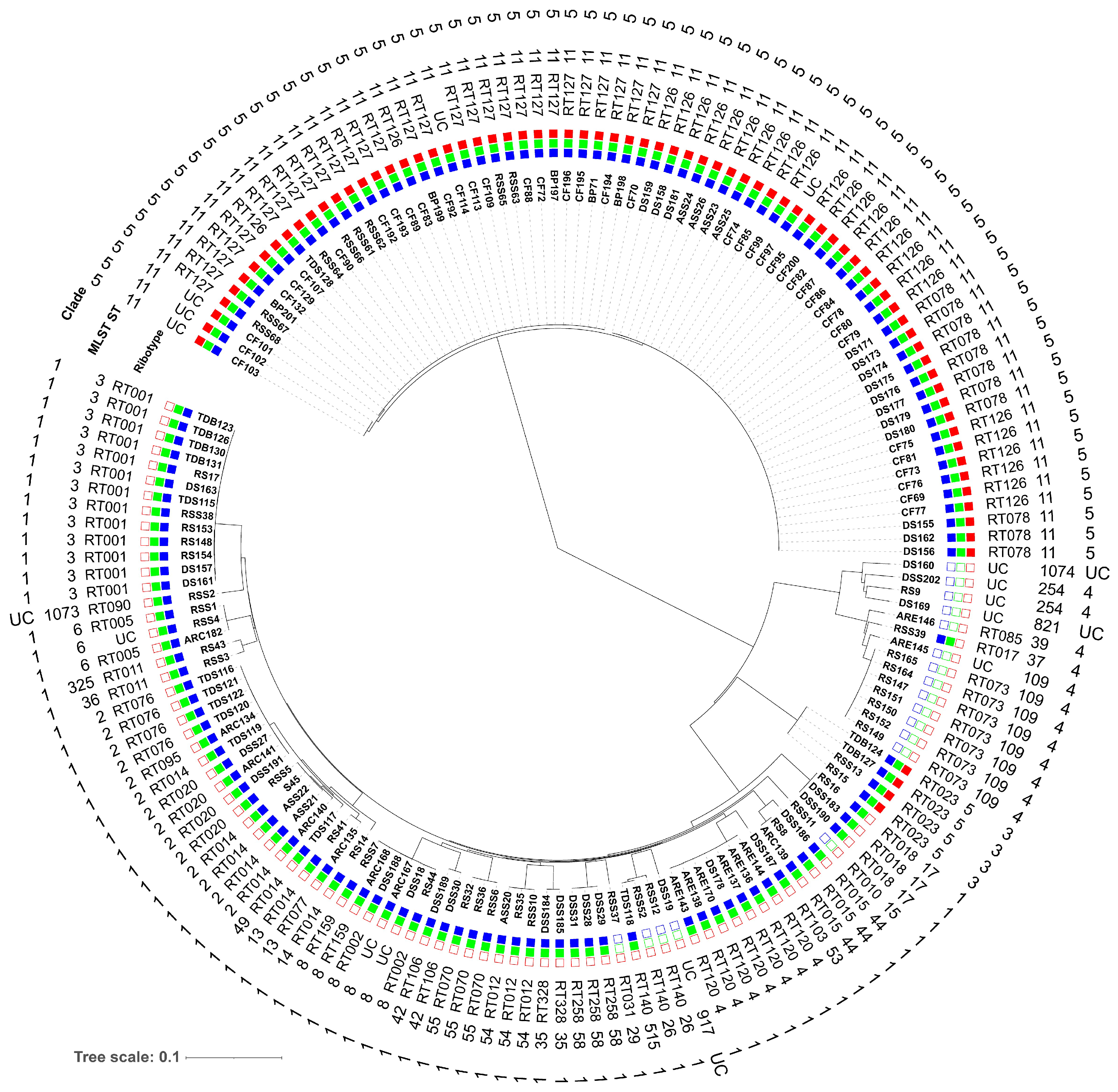
3.2. Molecular Subtyping, Molecular Epidemiology and Association with RTs and Toxin Genes
3.3. Antimicrobial Susceptibility
3.4. Antimicrobial Resistance (AMR) Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CDI | Clostridioides difficile infection |
| HA-CDI | Healthcare associated-CDI |
| CA-CDI | Community associated-CDI |
| RT | Ribotype |
| WGS | Whole-genome sequencing |
| ST | Sequence type |
| MLST | Multi-locus sequence typing |
| cgMLST | Core genome MLST |
| AMR | Antimicrobial resistance |
| HGT | Horizontal gene transfer |
| MGEs | Mobile genetic elements |
| MXF | Moxifloxacin |
| CLR | Clarithromycin |
| RIF | Rifampicin |
| MLSB | Macrolide-lincosamide-streptogramin B |
| RS | Raw sewage |
| ASS | Activated sewage sludge |
| RSS | Raw sewage sludge |
| DSS | Digested sewage sludge |
| TDS | Thermophilic digester for sewage sludge |
| TDB | Thermophilic digester for biowaste |
| ARC/E | Anaerobic lab scale bioreactors treating sewage sludge (control and experiment) |
| DSS-S | Digested sewage sludge-amended soils |
| WWTP | Wastewater treatment plant |
| CF | Calf feces |
| BP | Biogas plant digestate |
| S | Soil |
References
- Usacheva, E.A.; Jin, J.-P.; Peterson, L.R. Host response to Clostridium difficile infection: Diagnostics and detection. J. Glob. Antimicrob. Resist. 2016, 7, 93–101. [Google Scholar] [CrossRef]
- Buddle, J.E.; Fagan, R.P. Pathogenicity and virulence of Clostridioides difficile. Virulence 2023, 14, 2150452. [Google Scholar] [CrossRef]
- Peng, Z.; Jin, D.; Kim, H.B.; Stratton Charles, W.; Wu, B.; Tang, Y.-W.; Sun, X. Update on Antimicrobial Resistance in Clostridium difficile: Resistance Mechanisms and Antimicrobial Susceptibility Testing. J. Clin. Microbiol. 2017, 55, 1998–2008. [Google Scholar] [CrossRef]
- Spigaglia, P. Recent advances in the understanding of antibiotic resistance in Clostridium difficile infection. Ther. Adv. Infect. Dis. 2016, 3, 23–42. [Google Scholar] [CrossRef]
- O’Grady, K.; Knight, D.R.; Riley, T.V. Antimicrobial resistance in Clostridioides difficile. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 2459–2478. [Google Scholar] [CrossRef]
- Dingle, K.E.; Elliott, B.; Robinson, E.; Griffiths, D.; Eyre, D.W.; Stoesser, N.; Vaughan, A.; Golubchik, T.; Fawley, W.N.; Wilcox, M.H.; et al. Evolutionary history of the Clostridium difficile pathogenicity locus. Genome Biol. Evol. 2014, 6, 36–52. [Google Scholar] [CrossRef]
- Bolton, D.; Marcos, P. The Environment, Farm Animals and Foods as Sources of Clostridioides difficile Infection in Humans. Foods 2023, 12, 1094. [Google Scholar] [CrossRef]
- Lim, S.K.; Stuart, R.L.; Mackin, K.E.; Carter, G.P.; Kotsanas, D.; Francis, M.J.; Easton, M.; Dimovski, K.; Elliott, B.; Riley, T.V.; et al. Emergence of a ribotype 244 strain of Clostridium difficile associated with severe disease and related to the epidemic ribotype 027 strain. Clin. Infect. Dis. 2014, 58, 1723–1730. [Google Scholar] [CrossRef]
- Kuijper, E.J.; Barbut, F.; Brazier, J.S.; Kleinkauf, N.; Eckmanns, T.; Lambert, M.L.; Drudy, D.; Fitzpatrick, F.; Wiuff, C.; Brown, D.J.; et al. Update of Clostridium difficile infection due to PCR ribotype 027 in Europe, 2008. Eurosurveillance 2008, 13, 18942. [Google Scholar] [CrossRef]
- O’Connor, J.R.; Johnson, S.; Gerding, D.N. Clostridium difficile infection caused by the epidemic BI/NAP1/027 strain. Gastroenterology 2009, 136, 1913–1924. [Google Scholar] [CrossRef]
- Collins, D.A.; Hawkey, P.M.; Riley, T.V. Epidemiology of Clostridium difficile infection in Asia. Antimicrob. Resist. Infect. Control 2013, 2, 21. [Google Scholar] [CrossRef]
- Bauer, M.P.; Notermans, D.W.; van Benthem, B.H.B.; Brazier, J.S.; Wilcox, M.H.; Rupnik, M.; Monnet, D.L.; van Dissel, J.T.; Kuijper, E.J. Clostridium difficile infection in Europe: A hospital-based survey. Lancet 2011, 377, 63–73. [Google Scholar] [CrossRef]
- Goorhuis, A.; Bakker, D.; Corver, J.; Debast, S.B.; Harmanus, C.; Notermans, D.W.; Bergwerff, A.A.; Dekker, F.W.; Kuijper, E.J. Emergence of Clostridium difficile infection due to a new hypervirulent strain, polymerase chain reaction ribotype 078. Clin. Infect. Dis. 2008, 47, 1162–1170. [Google Scholar] [CrossRef]
- Tsai, B.-Y.; Chien, C.-C.; Huang, S.-H.; Zheng, J.-Y.; Hsu, C.-Y.; Tsai, Y.-S.; Hung, Y.-P.; Ko, W.-C.; Tsai, P.-J. The emergence of Clostridioides difficile PCR ribotype 127 at a hospital in northeastern Taiwan. J. Microbiol. Immunol. Infect. 2022, 55, 896–909. [Google Scholar] [CrossRef]
- Curova, K.; Novotny, M.; Ambro, L.; Kamlarova, A.; Lovayova, V.; Hrabovsky, V.; Siegfried, L.; Jarcuska, P.; Jarcuska, P.; Toporova, A. High Prevalence of Clostridioides difficile Ribotype 176 in the University Hospital in Kosice. Pathogens 2023, 12, 430. [Google Scholar] [CrossRef]
- Rabold, D.; Espelage, W.; Abu Sin, M.; Eckmanns, T.; Schneeberg, A.; Neubauer, H.; Möbius, N.; Hille, K.; Wieler, L.H.; Seyboldt, C.; et al. The zoonotic potential of Clostridium difficile from small companion animals and their owners. PLoS ONE 2018, 13, e0193411. [Google Scholar] [CrossRef]
- Rodriguez Diaz, C.; Seyboldt, C.; Rupnik, M. Non-human C. difficile Reservoirs and Sources: Animals, Food, Environment. Adv. Exp. Med. Biol. 2018, 1050, 227–243. [Google Scholar] [CrossRef]
- Knight, D.R.; Riley, T.V. Genomic Delineation of Zoonotic Origins of Clostridium difficile. Front. Public Health 2019, 7, 164. [Google Scholar] [CrossRef]
- Heise, J.; Witt, P.; Maneck, C.; Wichmann-Schauer, H.; Maurischat, S. Prevalence and phylogenetic relationship of Clostridioides difficile strains in fresh poultry meat samples processed in different cutting plants. Int. J. Food Microbiol. 2021, 339, 109032. [Google Scholar] [CrossRef]
- Frentrup, M.; Thiel, N.; Junker, V.; Behrens, W.; Münch, S.; Siller, P.; Kabelitz, T.; Faust, M.; Indra, A.; Baumgartner, S.; et al. Agricultural fertilization with poultry manure results in persistent environmental contamination with the pathogen Clostridioides difficile. Environ. Microbiol. 2021, 23, 7591–7602. [Google Scholar] [CrossRef]
- Blasi, F.; Lovito, C.; Albini, E.; Bano, L.; Dalmonte, G.; Drigo, I.; Maresca, C.; Massacci, F.R.; Orsini, S.; Primavilla, S.; et al. Clostridioides difficile in Calves in Central Italy: Prevalence, Molecular Typing, Antimicrobial Susceptibility and Association with Antibiotic Administration. Animals 2021, 11, 515. [Google Scholar] [CrossRef]
- Blau, K.; Gallert, C. Prevalence, Antimicrobial Resistance and Toxin-Encoding Genes of Clostridioides difficile from Environmental Sources Contaminated by Feces. Antibiotics 2023, 12, 162. [Google Scholar] [CrossRef]
- Eckert, C.; Burghoffer, B.; Barbut, F. Contamination of ready-to-eat raw vegetables with Clostridium difficile in France. J. Med. Microbiol. 2013, 62, 1435–1438. [Google Scholar] [CrossRef]
- Dharmasena, M.; Jiang, X. Isolation of Toxigenic Clostridium difficile from Animal Manure and Composts Being Used as Biological Soil Amendments. Appl. Environ. Microbiol. 2018, 84, e00738-18. [Google Scholar] [CrossRef]
- Xu, C.; Weese, J.S.; Flemming, C.; Odumeru, J.; Warriner, K. Fate of Clostridium difficile during wastewater treatment and incidence in Southern Ontario watersheds. J. Appl. Microbiol. 2014, 117, 891–904. [Google Scholar] [CrossRef]
- Álvarez-Pérez, S.; Blanco, J.L.; Peláez, T.; Astorga, R.J.; Harmanus, C.; Kuijper, E.; García, M.E. High prevalence of the epidemic Clostridium difficile PCR ribotype 078 in Iberian free-range pigs. Res. Vet. Sci. 2013, 95, 358–361. [Google Scholar] [CrossRef]
- Bakker, D.; Corver, J.; Harmanus, C.; Goorhuis, A.; Keessen, E.C.; Fawley, W.N.; Wilcox, M.H.; Kuijper, E.J. Relatedness of human and animal Clostridium difficile PCR ribotype 078 isolates determined on the basis of multilocus variable-number tandem-repeat analysis and tetracycline resistance. J. Clin. Microbiol. 2010, 48, 3744–3749. [Google Scholar] [CrossRef]
- Goorhuis, A.; Debast, S.B.; van Leengoed, L.A.M.G.; Harmanus, C.; Notermans, D.W.; Bergwerff, A.A.; Kuijper, E.J. Clostridium difficile PCR ribotype 078: An emerging strain in humans and in pigs? J. Clin. Microbiol. 2008, 46, 1157, 1158. [Google Scholar] [CrossRef]
- Martínez-Meléndez, A.; Morfin-Otero, R.; Villarreal-Treviño, L.; Baines, S.D.; Camacho-Ortíz, A.; Garza-González, E. Molecular epidemiology of predominant and emerging Clostridioides difficile ribotypes. J. Microbiol. Methods 2020, 175, 105974. [Google Scholar] [CrossRef]
- Brouwer, M.S.M.; Roberts, A.P.; Hussain, H.; Williams, R.J.; Allan, E.; Mullany, P. Horizontal gene transfer converts non-toxigenic Clostridium difficile strains into toxin producers. Nat. Commun. 2013, 4, 2601. [Google Scholar] [CrossRef]
- Mullany, P.; Wilks, M.; Lamb, I.; Clayton, C.; Wren, B.; Tabaqchali, S. Genetic analysis of a tetracycline resistance element from Clostridium difficile and its conjugal transfer to and from Bacillus subtilis. J. Gen. Microbiol. 1990, 136, 1343–1349. [Google Scholar] [CrossRef]
- Jasni, A.S.; Mullany, P.; Hussain, H.; Roberts, A.P. Demonstration of conjugative transposon (Tn5397)-mediated horizontal gene transfer between Clostridium difficile and Enterococcus faecalis. Antimicrob. Agents Chemother. 2010, 54, 4924–4926. [Google Scholar] [CrossRef]
- Lemee, L.; Dhalluin, A.; Testelin, S.; Mattrat, M.-A.; Maillard, K.; Lemeland, J.-F.; Pons, J.-L. Multiplex PCR targeting tpi (triose phosphate isomerase), tcdA (Toxin A), and tcdB (Toxin B) genes for toxigenic culture of Clostridium difficile. J. Clin. Microbiol. 2004, 42, 5710–5714. [Google Scholar] [CrossRef]
- Abdrabou, A.M.M.; Ul Habib Bajwa, Z.; Halfmann, A.; Mellmann, A.; Nimmesgern, A.; Margardt, L.; Bischoff, M.; von Müller, L.; Gärtner, B.; Berger, F.K. Molecular epidemiology and antimicrobial resistance of Clostridioides difficile in Germany, 2014-2019. Int. J. Med. Microbiol. 2021, 311, 151507. [Google Scholar] [CrossRef]
- Persson, S.; Torpdahl, M.; Olsen, K.E.P. New multiplex PCR method for the detection of Clostridium difficile toxin A (tcdA) and toxin B (tcdB) and the binary toxin (cdtA/cdtB) genes applied to a Danish strain collection. Clin. Microbiol. Infect. 2008, 14, 1057–1064. [Google Scholar] [CrossRef]
- van Almsick, V.; Schuler, F.; Mellmann, A.; Schwierzeck, V. The Use of Long-Read Sequencing Technologies in Infection Control: Horizontal Transfer of a bla(CTX-M-27) Containing lncFII Plasmid in a Patient Screening Sample. Microorganisms 2022, 10, 491. [Google Scholar] [CrossRef]
- Bletz, S.; Janezic, S.; Harmsen, D.; Rupnik, M.; Mellmann, A. Defining and Evaluating a Core Genome Multilocus Sequence Typing Scheme for Genome-Wide Typing of Clostridium difficile. J. Clin. Microbiol. 2018, 56, e01987-17. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Hua, X.; Liang, Q.; Deng, M.; He, J.; Wang, M.; Hong, W.; Wu, J.; Lu, B.; Leptihn, S.; Yu, Y.; et al. BacAnt: A Combination Annotation Server for Bacterial DNA Sequences to Identify Antibiotic Resistance Genes, Integrons, and Transposable Elements. Front. Microbiol. 2021, 12, 649969. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.S.; Roshan, P.B.; Diene, S.M.; Rafael, L.-R.; Marie, K.; Luce, L.; Jean-Marc, R. ARG-ANNOT, a New Bioinformatic Tool to Discover Antibiotic Resistance Genes in Bacterial Genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef]
- Wang, M.; Goh, Y.-X.; Tai, C.; Wang, H.; Deng, Z.; Ou, H.-Y. VRprofile2: Detection of antibiotic resistance-associated mobilome in bacterial pathogens. Nucleic Acids Res. 2022, 50, W768–W773. [Google Scholar] [CrossRef] [PubMed]
- Spigaglia, P.; Barbanti, F.; Faccini, S.; Vescovi, M.; Criscuolo, E.M.; Ceruti, R.; Gaspano, C.; Rosignoli, C. Clostridioides difficile in Pigs and Dairy Cattle in Northern Italy: Prevalence, Characterization and Comparison between Animal and Human Strains. Microorganisms 2023, 11, 1738. [Google Scholar] [CrossRef] [PubMed]
- Marcos, P.; Whyte, P.; Burgess, C.; Ekhlas, D.; Bolton, D. Detection and Genomic Characterisation of Clostridioides difficile from Spinach Fields. Pathogens 2022, 11, 1310. [Google Scholar] [CrossRef] [PubMed]
- Shivaperumal, N.; Chang, B.J.; Riley, T.V. High Prevalence of Clostridium difficile in Home Gardens in Western Australia. Appl. Environ. Microbiol. 2020, 87, e01572-20. [Google Scholar] [CrossRef] [PubMed]
- Knight, D.R.; Thean, S.; Putsathit, P.; Fenwick, S.; Riley, T.V. Cross-sectional study reveals high prevalence of Clostridium difficile non-PCR ribotype 078 strains in Australian veal calves at slaughter. Appl. Environ. Microbiol. 2013, 79, 2630–2635. [Google Scholar] [CrossRef] [PubMed]
- Andrés-Lasheras, S.; Bolea, R.; Mainar-Jaime, R.C.; Kuijper, E.; Sevilla, E.; Martín-Burriel, I.; Chirino-Trejo, M. Presence of Clostridium difficile in pig faecal samples and wild animal species associated with pig farms. J. Appl. Microbiol. 2017, 122, 462–472. [Google Scholar] [CrossRef]
- Zidaric, V.; Pardon, B.; Dos Vultos, T.; Deprez, P.; Brouwer, M.S.M.; Roberts, A.P.; Henriques, A.O.; Rupnik, M. Different antibiotic resistance and sporulation properties within multiclonal Clostridium difficile PCR ribotypes 078, 126, and 033 in a single calf farm. Appl. Environ. Microbiol. 2012, 78, 8515–8522. [Google Scholar] [CrossRef]
- Hung, Y.-P.; Lin, H.-J.; Tsai, B.-Y.; Liu, H.-C.; Liu, H.-C.; Lee, J.-C.; Wu, Y.-H.; Wilcox, M.H.; Fawley, W.N.; Hsueh, P.-R.; et al. Clostridium difficile ribotype 126 in southern Taiwan: A cluster of three symptomatic cases. Anaerobe 2014, 30, 188–192. [Google Scholar] [CrossRef]
- Primavilla, S.; Farneti, S.; Petruzzelli, A.; Drigo, I.; Scuota, S. Contamination of hospital food with Clostridium difficile in Central Italy. Anaerobe 2019, 55, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.C.; Reid-Smith, R.; Gow, S.; Hannon, S.J.; Booker, C.; Rousseau, J.; Benedict, K.M.; Morley, P.S.; Weese, J.S. Prevalence and molecular characterization of Clostridium difficile isolated from feedlot beef cattle upon arrival and mid-feeding period. BMC Vet. Res. 2012, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Hammitt, M.C.; Bueschel, D.M.; Keel, M.K.; Glock, R.D.; Cuneo, P.; DeYoung, D.W.; Reggiardo, C.; Trinh, H.T.; Songer, J.G. A possible role for Clostridium difficile in the etiology of calf enteritis. Vet. Microbiol. 2008, 127, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I.; Borah, P.; Sharma, R.K.; Rajkhowa, S.; Rupnik, M.; Saikia, D.P.; Hasin, D.; Hussain, I.; Deka, N.K.; Barkalita, L.M.; et al. Molecular characteristics of Clostridium difficile isolates from human and animals in the North Eastern region of India. Mol. Cell. Probes 2016, 30, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-J.; Yang, L.; Gu, X.-X.; Chen, P.-X.; Fu, J.-L.; Jiang, H.-X. The first isolation of Clostridium difficile RT078/ST11 from pigs in China. PLoS ONE 2019, 14, e0212965. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.; Vernon, J.; Pilling, S.; Morris, K.; Nicolson, S.; Shearman, S.; Clark, E.; Palacios-Fabrega, J.A.; Wilcox, M. Five-year Pan-European, longitudinal surveillance of Clostridium difficile ribotype prevalence and antimicrobial resistance: The extended ClosER study. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Berger, F.K.; Gfrörer, S.; Becker, S.L.; Baldan, R.; Cirillo, D.M.; Frentrup, M.; Steglich, M.; Engling, P.; Nübel, U.; Mellmann, A.; et al. Hospital outbreak due to Clostridium difficile ribotype 018 (RT018) in Southern Germany. Int. J. Med. Microbiol. 2019, 309, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Baldan, R.; Trovato, A.; Bianchini, V.; Biancardi, A.; Cichero, P.; Mazzotti, M.; Nizzero, P.; Moro, M.; Ossi, C.; Scarpellini, P.; et al. Clostridium difficile PCR Ribotype 018, a Successful Epidemic Genotype. J. Clin. Microbiol. 2015, 53, 2575–2580. [Google Scholar] [CrossRef]
- Janezic, S.; Ocepek, M.; Zidaric, V.; Rupnik, M. Clostridium difficile genotypes other than ribotype 078 that are prevalent among human, animal and environmental isolates. BMC Microbiol. 2012, 12, 48. [Google Scholar] [CrossRef]
- Okada, Y.; Yagihara, Y.; Wakabayashi, Y.; Igawa, G.; Saito, R.; Higurashi, Y.; Ikeda, M.; Tatsuno, K.; Okugawa, S.; Moriya, K. Epidemiology and virulence-associated genes of Clostridioides difficile isolates and factors associated with toxin EIA results at a university hospital in Japan. Access Microbiol. 2020, 2, acmi000086. [Google Scholar] [CrossRef]
- Baktash, A.; Corver, J.; Harmanus, C.; Smits, W.K.; Fawley, W.; Wilcox, M.H.; Kumar, N.; Eyre, D.W.; Indra, A.; Mellmann, A.; et al. Comparison of Whole-Genome Sequence-Based Methods and PCR Ribotyping for Subtyping of Clostridioides difficile. J. Clin. Microbiol. 2022, 60, e0173721. [Google Scholar] [CrossRef] [PubMed]
- Frentrup, M.; Zhou, Z.; Steglich, M.; Meier-Kolthoff, J.P.; Göker, M.; Riedel, T.; Bunk, B.; Spröer, C.; Overmann, J.; Blaschitz, M.; et al. A publicly accessible database for Clostridioides difficile genome sequences supports tracing of transmission chains and epidemics. Microb. Genom. 2020, 6, mgen000410. [Google Scholar] [CrossRef] [PubMed]
- Janezic, S.; Rupnik, M. Genomic diversity of Clostridium difficile strains. Res. Microbiol. 2015, 166, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Knight, D.R.; Elliott, B.; Chang, B.J.; Perkins, T.T.; Riley, T.V. Diversity and Evolution in the Genome of Clostridium difficile. Clin. Microbiol. Rev. 2015, 28, 721–741. [Google Scholar] [CrossRef] [PubMed]
- Imwattana, K.; Kiratisin, P.; Riley, T.V.; Knight, D.R. Genomic basis of antimicrobial resistance in non-toxigenic Clostridium difficile in Southeast Asia. Anaerobe 2020, 66, 102290. [Google Scholar] [CrossRef] [PubMed]
- Dingle, K.E.; Didelot, X.; Quan, T.P.; Eyre, D.W.; Stoesser, N.; Marwick, C.A.; Coia, J.; Brown, D.; Buchanan, S.; Ijaz, U.Z.; et al. A Role for Tetracycline Selection in Recent Evolution of Agriculture-Associated Clostridium difficile PCR Ribotype 078. mBio 2019, 10, e02790-18. [Google Scholar] [CrossRef] [PubMed]
- Sloan, J.; McMurry, L.M.; Lyras, D.; Levy, S.B.; Rood, J.I. The Clostridium perfringens Tet P determinant comprises two overlapping genes: tetA(P), which mediates active tetracycline efflux, and tetB(P), which is related to the ribosomal protection family of tetracycline-resistance determinants. Mol. Microbiol. 1994, 11, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Spigaglia, P.; Mastrantonio, P.; Barbanti, F. Antibiotic Resistances of Clostridium difficile. Adv. Exp. Med. Biol. 2018, 1050, 137–159. [Google Scholar] [CrossRef] [PubMed]
- Werner, G.; Hildebrandt, B.; Witte, W. Aminoglycoside-streptothricin resistance gene cluster aadE-sat4-aphA-3 disseminated among multiresistant isolates of Enterococcus faecium. Antimicrob. Agents Chemother. 2001, 45, 3267–3269. [Google Scholar] [CrossRef]
- Jacob, J.; Evers, S.; Bischoff, K.; Carlier, C.; Courvalin, P. Characterization of the sat4 gene encoding a streptothricin acetyltransferase in Campylobacter coli BE/G4. FEMS Microbiol. Lett. 1994, 120, 13–17. [Google Scholar] [CrossRef]
- Spigaglia, P.; Barbanti, F.; Mastrantonio, P. Multidrug resistance in European Clostridium difficile clinical isolates. J. Antimicrob. Chemother. 2011, 66, 2227–2234. [Google Scholar] [CrossRef]
- Eubank, T.A.; Gonzales-Luna, A.J.; Hurdle, J.G.; Garey, K.W. Genetic Mechanisms of Vancomycin Resistance in Clostridioides difficile: A Systematic Review. Antibiotics 2022, 11, 258. [Google Scholar] [CrossRef]
- Knight, D.R.; Riley, T.V. Clostridium difficile clade 5 in Australia: Antimicrobial susceptibility profiling of PCR ribotypes of human and animal origin. J. Antimicrob. Chemother. 2016, 71, 2213–2217. [Google Scholar] [CrossRef]
- Suzuki, H.; Tomita, M.; Tsai, P.-J.; Ko, W.-C.; Hung, Y.-P.; Huang, I.-H.; Chen, J.-W. Comparative genomic analysis of Clostridium difficile ribotype 027 strains including the newly sequenced strain NCKUH-21 isolated from a patient in Taiwan. Gut Pathog. 2017, 9, 70. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clade | RT | ST | Clade | RT | ST |
|---|---|---|---|---|---|
| Clade 1 | RT005 * | ST6 | Clade 1 | RT031 | ST29 |
| RT090 | ST1073 | RT001 * | ST3 | ||
| RT011 * | ST36, ST325 | RT015 * | ST44 | ||
| RT020 * | ST2 | RT014 * | ST14, ST13, ST2, ST49 | ||
| RT070 * | ST55 | RT018 * | ST17 | ||
| RT159 | ST8 | RT002 * | ST8 | ||
| RT012 * | ST54 | RT258 * | ST58 | ||
| RT010 | ST15 | RT103 * | ST53 | ||
| RT140 | ST26, ST515 | Clade 4 | RT085 * | ST39 | |
| RT077 * | ST13 | RT017 * | ST37 | ||
| RT328 * | ST35 | RT073 | ST109 | ||
| RT106 * | ST42 | Clade 5 | RT126 * | ST11 | |
| RT076 * | ST2 | RT127 * | |||
| RT095 | ST2 | RT078 * | |||
| RT120 | ST4 | Clade 3 | RT023 * | ST5 |
| RT/ST | No. of Isolates (%) | ||
|---|---|---|---|
| CLR | MXF | RIF | |
| RT126/ST11 | 24 (89%) | 11 (41%) | 1 (4%) |
| RT078/ST11 | 4 (36%) | 5 (45%) | 0 |
| RT001/ST3 | 2 (15%) | 2 (15%) | 0 |
| RT012/ST54 | 3 (100%) | 0 | 0 |
| RT140/ST26/ST515 | 2 (67%) | 2 (67%) | 0 |
| RT328/ST35 | 2 (100%) | 0 | 0 |
| RT010/ST15 | 1 (100% | 0 | 0 |
| RT031/ST29 | 1 (100%) | 0 | 0 |
| RT017/ST37 | 0 | 1 (100%) | 1 (100%) |
| RT106/ST42 | 1 (50%) | 0 | 0 |
| RT015/ST44 | 0 | 1 (33%) | 0 |
| RT014/ST2 | 1 (13%) | 0 | 0 |
| RT085/ST39 | 1 (100%) | 0 | 1 (100%) |
| UC/ST11 | 1 (20%) | 1 (20%) | 0 |
| Total | 43 (26%) | 23 (14%) | 3 (2%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blau, K.; Berger, F.K.; Mellmann, A.; Gallert, C. Clostridioides difficile from Fecally Contaminated Environmental Sources: Resistance and Genetic Relatedness from a Molecular Epidemiological Perspective. Microorganisms 2023, 11, 2497. https://doi.org/10.3390/microorganisms11102497
Blau K, Berger FK, Mellmann A, Gallert C. Clostridioides difficile from Fecally Contaminated Environmental Sources: Resistance and Genetic Relatedness from a Molecular Epidemiological Perspective. Microorganisms. 2023; 11(10):2497. https://doi.org/10.3390/microorganisms11102497
Chicago/Turabian StyleBlau, Khald, Fabian K. Berger, Alexander Mellmann, and Claudia Gallert. 2023. "Clostridioides difficile from Fecally Contaminated Environmental Sources: Resistance and Genetic Relatedness from a Molecular Epidemiological Perspective" Microorganisms 11, no. 10: 2497. https://doi.org/10.3390/microorganisms11102497