
Bacterial Biomarkers of the Oropharyngeal and Oral Cavity during SARS-CoV-2 Infection

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Samples from the Quebec Biobank of COVID-19
2.3. DNA Extraction
2.4. Library Preparation and Sequencing
2.5. Sequence Processing
2.6. Diversity Analyses
2.7. Community Composition across Positive Test (PT) and Negative Test (NT) Groups
2.8. Abundance of Specific Taxa in the Positive Test (PT) and Negative Test (NT) Groups
3. Results
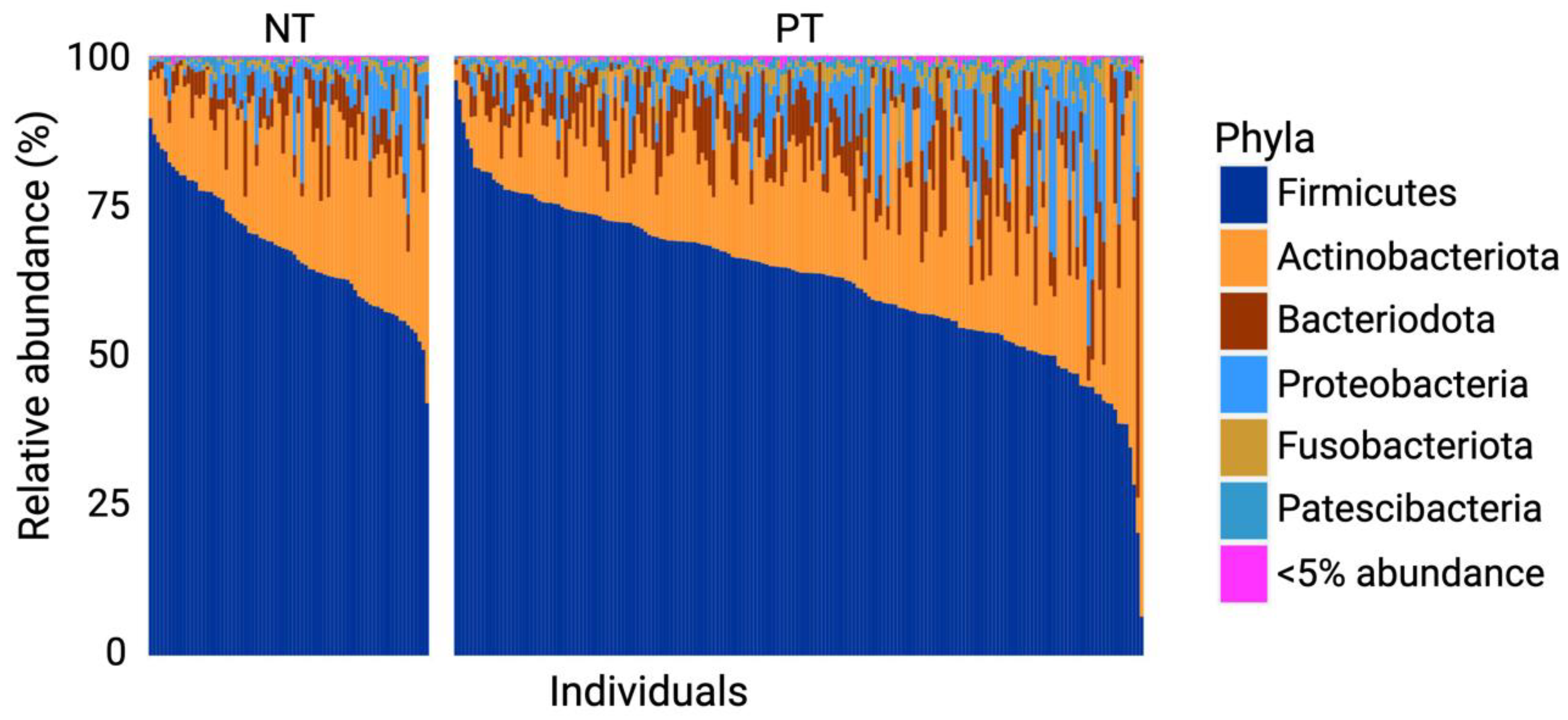
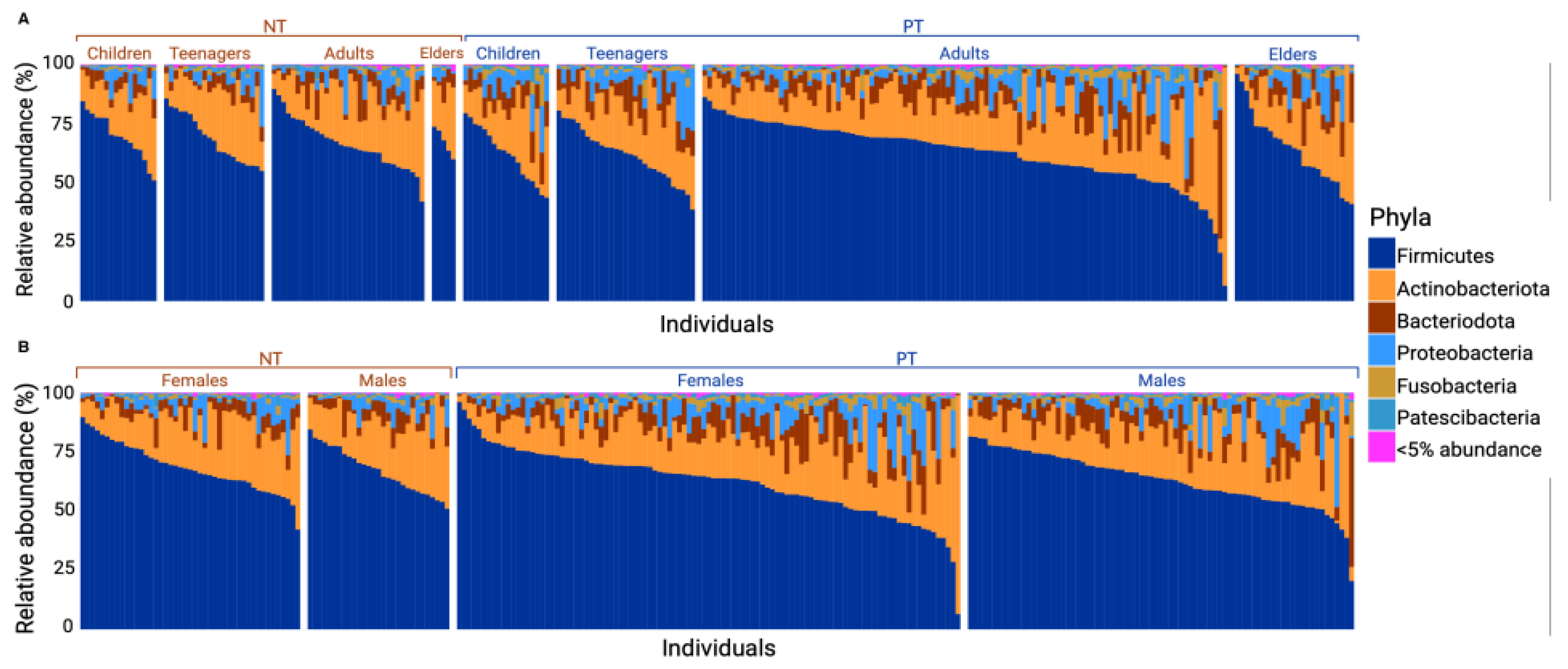
3.1. Firmicutes and Actinobacteriota Dominate the OCO Microbiome
3.2. No Significant Difference in Alpha Diversity between PT and NT Individuals
3.3. Significant Difference in Beta Diversity in Bacterial Communities in Samples of the Positive Test (PT) and Negative Test (NT) Groups
3.4. Prevotella and Veillonella Are Features of OCO in COVID-19 Patients
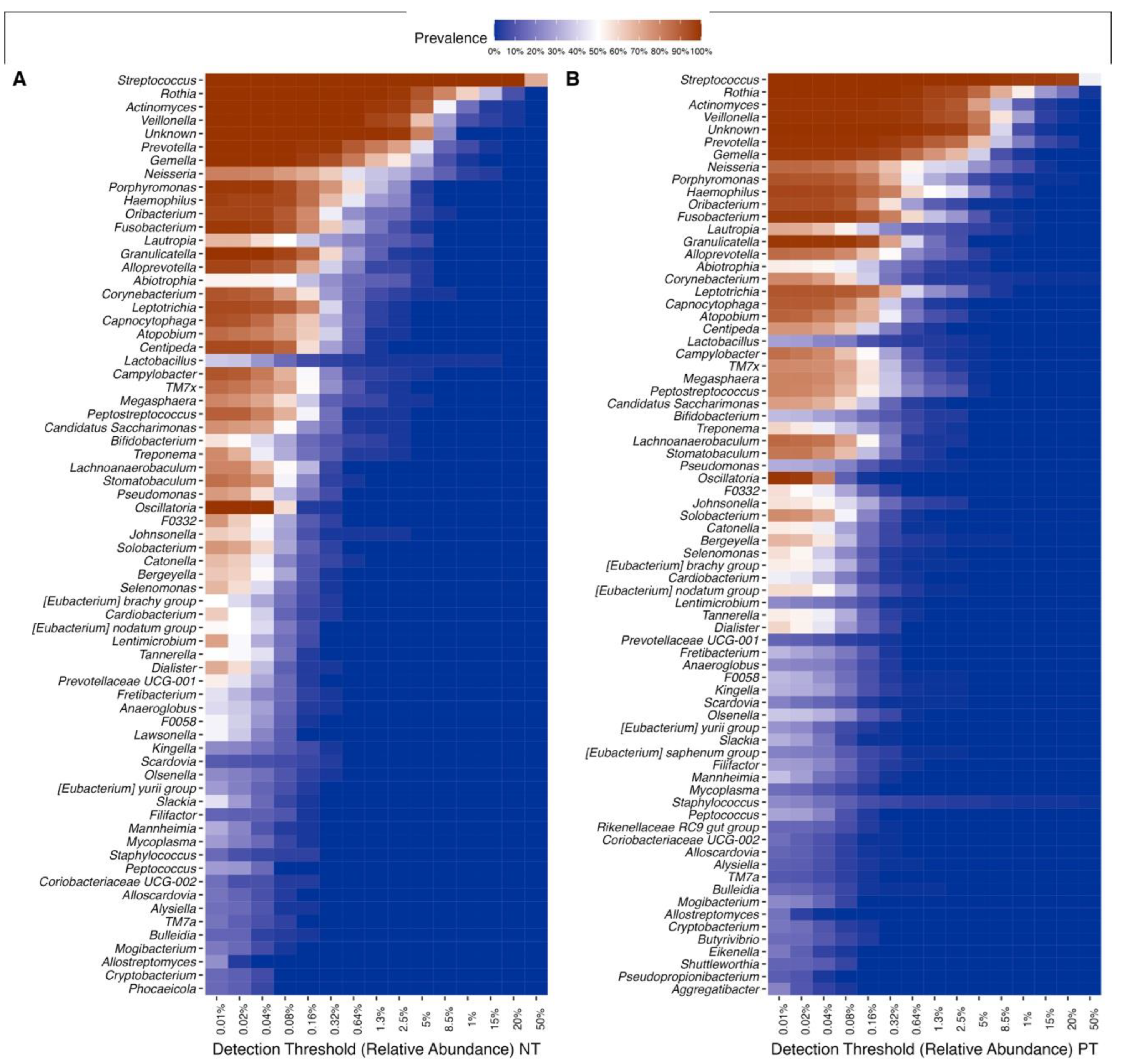
3.5. Individuals with COVID-19 Exhibit Greater Diversity within Their Core OCO Microbiome
3.6. COVID-19 Infection Status Is Associated with Differentially Abundant Taxa
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. World Health Organization Coronavirus Disease (COVID-19) Pandemic 2022; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- World Health Organization. World Health Organization WHO Coronavirus (COVID-19) Dashboard 2022; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Sun, P.; Lu, X.; Xu, C.; Sun, W.; Pan, B. Understanding of COVID-19 Based on Current Evidence. J. Med. Virol. 2020, 92, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Leon, S.; Wegman-Ostrosky, T.; Perelman, C.; Sepulveda, R.; Rebolledo, P.A.; Cuapio, A.; Villapol, S. More than 50 Long-Term Effects of COVID-19: A Systematic Review and Meta-Analysis. Sci. Rep. 2021, 11, 16144. [Google Scholar] [CrossRef] [PubMed]
- Gavriatopoulou, M.; Ntanasis-Stathopoulos, I.; Korompoki, E.; Fotiou, D.; Migkou, M.; Tzanninis, I.-G.; Psaltopoulou, T.; Kastritis, E.; Terpos, E.; Dimopoulos, M.A. Emerging Treatment Strategies for COVID-19 Infection. Clin. Exp. Med. 2021, 21, 167–179. [Google Scholar] [CrossRef]
- Brown, R.L.; Sequeira, R.P.; Clarke, T.B. The Microbiota Protects against Respiratory Infection via GM-CSF Signaling. Nat. Commun. 2017, 8, 1512. [Google Scholar] [CrossRef]
- Man, W.H.; de Steenhuijsen Piters, W.A.A.; Bogaert, D. The Microbiota of the Respiratory Tract: Gatekeeper to Respiratory Health. Nat. Rev. Microbiol. 2017, 15, 259–270. [Google Scholar] [CrossRef]
- Gollwitzer, E.S.; Saglani, S.; Trompette, A.; Yadava, K.; Sherburn, R.; McCoy, K.D.; Nicod, L.P.; Lloyd, C.M.; Marsland, B.J. Lung Microbiota Promotes Tolerance to Allergens in Neonates via PD-L1. Nat. Med. 2014, 20, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Porto, B.N.; Moraes, T.J. The Triad: Respiratory Microbiome–Virus–Immune Response in the Pathophysiology of Pulmonary Viral Infections. Expert Rev. Respir. Med. 2021, 15, 635–648. [Google Scholar] [CrossRef]
- Ichinohe, T.; Pang, I.K.; Kumamoto, Y.; Peaper, D.R.; Ho, J.H.; Murray, T.S.; Iwasaki, A. Microbiota Regulates Immune Defense against Respiratory Tract Influenza A Virus Infection. Proc. Natl. Acad. Sci. USA 2011, 108, 5354–5359. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, F.; Zhou, F.; Li, H.; Ge, W.; Gan, R.; Nie, H.; Li, B.; Wang, Y.; Wu, M. Metagenomic Analysis Reveals Oropharyngeal Microbiota Alterations in Patients with COVID-19. Signal Transduct. Target. Ther. 2021, 6, 191. [Google Scholar] [CrossRef]
- Molyneaux, P.L.; Cox, M.J.; Willis-Owen, S.A.G.; Mallia, P.; Russell, K.E.; Russell, A.-M.; Murphy, E.; Johnston, S.L.; Schwartz, D.A.; Wells, A.U.; et al. The Role of Bacteria in the Pathogenesis and Progression of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 906–913. [Google Scholar] [CrossRef]
- de Steenhuijsen Piters, W.A.A.; Sanders, E.A.M.; Bogaert, D. The Role of the Local Microbial Ecosystem in Respiratory Health and Disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140294. [Google Scholar] [CrossRef] [PubMed]
- Pichon, M.; Lina, B.; Josset, L. Impact of the Respiratory Microbiome on Host Responses to Respiratory Viral Infection. Vaccines 2017, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Wypych, T.P.; Wickramasinghe, L.C.; Marsland, B.J. The Influence of the Microbiome on Respiratory Health. Nat. Immunol. 2019, 20, 1279–1290. [Google Scholar] [CrossRef]
- Shahbazi, R.; Yasavoli-Sharahi, H.; Alsadi, N.; Ismail, N.; Matar, C. Probiotics in Treatment of Viral Respiratory Infections and Neuroinflammatory Disorders. Molecules 2020, 25, 4891. [Google Scholar] [CrossRef] [PubMed]
- Sudo, N.; Yu, X.; Aiba, Y.; Oyama, N.; Sonoda, J.; Koga, Y.; Kubo, C. An Oral Introduction of Intestinal Bacteria Prevents the Development of a Long-term Th2-skewed Immunological Memory Induced by Neonatal Antibiotic Treatment in Mice. Clin. Exp. Allergy 2002, 32, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What Is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar]
- Jia, Y.; He, T.; Wu, D.; Tong, J.; Zhu, J.; Li, Z.; Dong, J. The Treatment of Qibai Pingfei Capsule on Chronic Obstructive Pulmonary Disease May Be Mediated by Th17/Treg Balance and Gut-Lung Axis Microbiota. J. Transl. Med. 2022, 20, 281. [Google Scholar] [CrossRef]
- Alharris, E.; Mohammed, A.; Alghetaa, H.; Zhou, J.; Nagarkatti, M.; Nagarkatti, P. The Ability of Resveratrol to Attenuate Ovalbumin-Mediated Allergic Asthma Is Associated with Changes in Microbiota Involving the Gut-Lung Axis, Enhanced Barrier Function and Decreased Inflammation in the Lungs. Front. Immunol. 2022, 13, 805770. [Google Scholar] [CrossRef]
- Yuksel, N.; Gelmez, B.; Yildiz-Pekoz, A. Lung Microbiota: Its Relationship to Respiratory System Diseases and Approaches for Lung-Targeted Probiotic Bacteria Delivery. Mol. Pharm. 2023, 20, 3320–3337. [Google Scholar] [CrossRef]
- Bassis, C.M.; Erb-Downward, J.R.; Dickson, R.P.; Freeman, C.M.; Schmidt, T.M.; Young, V.B.; Beck, J.M.; Curtis, J.L.; Huffnagle, G.B. Analysis of the Upper Respiratory Tract Microbiotas as the Source of the Lung and Gastric Microbiotas in Healthy Individuals. MBio 2015, 6, e00037-15. [Google Scholar] [CrossRef]
- Merenstein, C.; Liang, G.; Whiteside, S.A.; Cobián-Güemes, A.G.; Merlino, M.S.; Taylor, L.J.; Glascock, A.; Bittinger, K.; Tanes, C.; Graham-Wooten, J. Signatures of COVID-19 Severity and Immune Response in the Respiratory Tract Microbiome. MBio 2021, 12, e01777-21. [Google Scholar] [CrossRef] [PubMed]
- Haran, J.P.; Bradley, E.; Zeamer, A.L.; Cincotta, L.; Salive, M.-C.; Dutta, P.; Mutaawe, S.; Anya, O.; Meza-Segura, M.; Moormann, A.M. Inflammation-Type Dysbiosis of the Oral Microbiome Associates with the Duration of COVID-19 Symptoms and Long COVID. JCI Insight 2021, 6, e152346. [Google Scholar] [CrossRef] [PubMed]
- Bradley, E.S.; Zeamer, A.L.; Bucci, V.; Cincotta, L.; Salive, M.-C.; Dutta, P.; Mutaawe, S.; Anya, O.; Tocci, C.; Moormann, A. Oropharyngeal Microbiome Profiled at Admission Is Predictive of the Need for Respiratory Support Among COVID-19 Patients. medRxiv 2022, 13, 1009440. [Google Scholar] [CrossRef] [PubMed]
- Bchetnia, M.; Bouchard, L.; Mathieu, J.; Campeau, P.M.; Morin, C.; Brisson, D.; Laberge, A.-M.; Vézina, H.; Gaudet, D.; Laprise, C. Genetic Burden Linked to Founder Effects in Saguenay–Lac-Saint-Jean Illustrates the Importance of Genetic Screening Test Availability. J. Med. Genet. 2021, 58, 653–665. [Google Scholar] [CrossRef]
- Shifman, S.; Darvasi, A. The Value of Isolated Populations. Nat. Genet. 2001, 28, 309–310. [Google Scholar] [CrossRef]
- Chen, Z.; Azman, A.S.; Chen, X.; Zou, J.; Tian, Y.; Sun, R.; Xu, X.; Wu, Y.; Lu, W.; Ge, S. Global Landscape of SARS-CoV-2 Genomic Surveillance and Data Sharing. Nat. Genet. 2022, 54, 499–507. [Google Scholar] [CrossRef]
- CIUSSSMCQ. Méthode de Prélèvement Par Gargarisme Pour Le Diagnostic de La COVID-19. Available online: https://ciusssmcq.ca/Content/Client/Librairie/Documents/COVID-19/Personnel/Depistage/AFF_8_5X11_Depistage_Gargarisme_2.pdf (accessed on 15 June 2023).
- Tremblay, K.; Rousseau, S.; Zawati MN, H.; Auld, D.; Chassé, M.; Coderre, D.; Falcone, E.L.; Gauthier, N.; Grandvaux, N.; Gros-Louis, F. The Biobanque Québécoise de La COVID-19 (BQC19)—A Cohort to Prospectively Study the Clinical and Biological Determinants of COVID-19 Clinical Trajectories. PLoS ONE 2021, 16, e0245031. [Google Scholar]
- Statistique Canada. Catégories d’âge – groupes établis selon le cycle de vie. Available online: https://www.statcan.gc.ca/fr/concepts/definitions/age2 (accessed on 12 November 2022).
- ThermoFisher Scientific. ThermoFischer Scientific QubitTM 1X dsDNA High Sensitivity (HS) and Broad Range (BR) Assay Kits. Available online: https://www.thermofisher.com/order/catalog/product/Q33230 (accessed on 15 July 2023).
- Ciceri, F.; Castagna, A.; Rovere-Querini, P.; De Cobelli, F.; Ruggeri, A.; Galli, L.; Conte, C.; De Lorenzo, R.; Poli, A.; Ambrosio, A. Early Predictors of Clinical Outcomes of COVID-19 Outbreak in Milan, Italy. Clin. Immunol. 2020, 217, 108509. [Google Scholar] [CrossRef]
- Illumina. Illumina 16S Metagenomic Sequencing Library Preparation. Available online: https://support.illumina.com/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf (accessed on 15 June 2023).
- ThermoFisher Scientific. Invitrogen Quant-iTTM 1X dsDNA, high sensitivity (HS) Assay Kit. Available online: https://www.thermofisher.com/order/catalog/product/Q33232 (accessed on 15 June 2023).
- Allaire, J. RStudio: Integrated Development Environment for R. Boston MA 2012, 770, 165–171. [Google Scholar]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Callahan, B.; McMurdie, P.; Holmes, S. Package ‘dada2’. Available online: http://bioconductor.jp/packages/devel/bioc/manuals/dada2/man/dada2.pdf (accessed on 12 June 2022).
- Pagès, H. Package ‘Biostrings’. Available online: https://bioconductor.org/packages/release/bioc/manuals/Biostrings/man/Biostrings.pdf (accessed on 12 June 2022).
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple Statistical Identification and Removal of Contaminant Sequences in Marker-Gene and Metagenomics Data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Basic Storage, Access, and Manipulation of Phylogenetic Sequencing Data with Phyloseq. Available online: https://www.bioconductor.org/packages/devel/bioc/vignettes/phyloseq/inst/doc/phyloseq-basics.html (accessed on 3 July 2022).
- Shetty, S.A.; Lahti, L. Microbiomeutilities: An R Package for Utilities to Guide in-Depth Marker Gene Amplicon Data Analysis. Available online: https://doi.org/10.5281/zenodo.1471685 (accessed on 17 July 2022).
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Stevens, M.H.H.; Oksanen, M.J.; Suggests, M. The Vegan Package. Community Ecol. Package 2007, 10, 719. [Google Scholar]
- Cao, Y.; Dong, Q.; Wang, D.; Zhang, P.; Liu, Y.; Niu, C. microbiomeMarker: An R/Bioconductor Package for Microbiome Marker Identification and Visualization. Bioinformatics 2022, 38, 4027–4029. [Google Scholar] [CrossRef] [PubMed]
- Barnett, D.J.; Arts, I.C.; Penders, J. microViz: An R Package for Microbiome Data Visualization and Statistics. J. Open Source Softw. 2021, 6, 3201. [Google Scholar] [CrossRef]
- Lin, H.; Peddada, S.D. Analysis of composition of microbiomes with bias correction. Nat Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Wu, Y.; Cheng, X.; Jiang, G.; Tang, H.; Ming, S.; Tang, L.; Lu, J.; Guo, C.; Shan, H.; Huang, X. Altered Oral and Gut Microbiota and Its Association with SARS-CoV-2 Viral Load in COVID-19 Patients during Hospitalization. Npj Biofilms Microbiomes 2021, 7, 61. [Google Scholar] [CrossRef]
- Long, Q.-X.; Liu, B.-Z.; Deng, H.-J.; Wu, G.-C.; Deng, K.; Chen, Y.-K.; Liao, P.; Qiu, J.-F.; Lin, Y.; Cai, X.-F. Antibody Responses to SARS-CoV-2 in Patients with COVID-19. Nat. Med. 2020, 26, 845–848. [Google Scholar] [CrossRef]
- De Maio, F.; Posteraro, B.; Ponziani, F.R.; Cattani, P.; Gasbarrini, A.; Sanguinetti, M. Nasopharyngeal Microbiota Profiling of SARS-CoV-2 Infected Patients. Biol. Proced. Online 2020, 22, 18. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human Gut Microbiome Viewed across Age and Geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Thorsen, J.; Stokholm, J.; Rasmussen, M.A.; Roggenbuck-Wedemeyer, M.; Vissing, N.H.; Mortensen, M.S.; Brejnrod, A.D.; Fleming, L.; Bush, A.; Roberts, G.; et al. Asthma and Wheeze Severity and the Oropharyngeal Microbiota in Children and Adolescents. Ann. Am. Thorac. Soc. 2022, 19, 2031–2043. [Google Scholar] [CrossRef] [PubMed]
- Ihekweazu, F.D.; Versalovic, J. Development of the Pediatric Gut Microbiome: Impact on Health and Disease. Am. J. Med. Sci. 2018, 356, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Dickson, R.P.; Erb-Downward, J.R.; Martinez, F.J.; Huffnagle, G.B. The Microbiome and the Respiratory Tract. Annu. Rev. Physiol. 2016, 78, 481–504. [Google Scholar] [CrossRef] [PubMed]
- Rattanaburi, S.; Sawaswong, V.; Chitcharoen, S.; Sivapornnukul, P.; Nimsamer, P.; Suntronwong, N.; Puenpa, J.; Poovorawan, Y.; Payungporn, S. Bacterial Microbiota in Upper Respiratory Tract of COVID-19 and Influenza Patients. Exp. Biol. Med. 2022, 247, 409–415. [Google Scholar] [CrossRef]
- Bingula, R.; Filaire, M.; Radosevic-Robin, N.; Bey, M.; Berthon, J.-Y.; Bernalier-Donadille, A.; Vasson, M.-P.; Filaire, E. Desired Turbulence? Gut-Lung Axis, Immunity, and Lung Cancer. J. Oncol. 2017, 2017, 5035371. [Google Scholar]
- Lemon, K.P.; Klepac-Ceraj, V.; Schiffer, H.K.; Brodie, E.L.; Lynch, S.V.; Kolter, R. Comparative Analyses of the Bacterial Microbiota of the Human Nostril and Oropharynx. MBio 2010, 1, e00129-10. [Google Scholar] [CrossRef]
- Woodall, C.A.; McGeoch, L.J.; Hay, A.D.; Hammond, A. Respiratory Tract Infections and Gut Microbiome Modifications: A Systematic Review. PLoS ONE 2022, 17, e0262057. [Google Scholar] [CrossRef]
- Chattopadhyay, I.; Verma, M.; Panda, M. Role of Oral Microbiome Signatures in Diagnosis and Prognosis of Oral Cancer. Technol. Cancer Res. Treat. 2019, 18, 1533033819867354. [Google Scholar] [CrossRef]
- Nardelli, C.; Gentile, I.; Setaro, M.; Di Domenico, C.; Pinchera, B.; Buonomo, A.R.; Zappulo, E.; Scotto, R.; Scaglione, G.L.; Castaldo, G. Nasopharyngeal Microbiome Signature in COVID-19 Positive Patients: Can We Definitively Get a Role to Fusobacterium Periodonticum? Front. Cell Infect. Microbiol. 2021, 11, 625581. [Google Scholar] [CrossRef]
- Teixeira, P.C.; Dorneles, G.P.; Santana Filho, P.C.; da Silva, I.M.; Schipper, L.L.; Postiga, I.A.; Neves, C.A.M.; Junior, L.C.R.; Peres, A.; de Souto, J.T. Increased LPS Levels Coexist with Systemic Inflammation and Result in Monocyte Activation in Severe COVID-19 Patients. Int. Immunopharmacol. 2021, 100, 108125. [Google Scholar] [CrossRef]
- Bayston, K.F.; Cohen, J. Bacterial Endotoxin and Current Concepts in the Diagnosis and Treatment of Endotoxaemia. J. Med. Microbiol. 1990, 31, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Terán, A.; Mejía-Nepomuceno, F.; Herrera, M.T.; Barreto, O.; García, E.; Castillejos, M.; Boukadida, C.; Matias-Florentino, M.; Rincón-Rubio, A.; Avila-Rios, S. Dysbiosis and Structural Disruption of the Respiratory Microbiota in COVID-19 Patients with Severe and Fatal Outcomes. Sci. Rep. 2021, 11, 21297. [Google Scholar] [CrossRef] [PubMed]
- Ventero, M.P.; Cuadrat, R.R.; Vidal, I.; Andrade, B.G.; Molina-Pardines, C.; Haro-Moreno, J.M.; Coutinho, F.H.; Merino, E.; Regitano, L.C.; Silveira, C.B. Nasopharyngeal Microbial Communities of Patients Infected with SARS-CoV-2 That Developed COVID-19. Front. Microbiol. 2021, 12, 560. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Salazar, C.; Kimura, K.S.; Shilts, M.H.; Strickland, B.A.; Freeman, M.H.; Wessinger, B.C.; Gupta, V.; Brown, H.M.; Rajagopala, S.V.; Turner, J.H. SARS-CoV-2 Infection and Viral Load Are Associated with the Upper Respiratory Tract Microbiome. J. Allergy Clin. Immunol. 2021, 147, 1226–1233. [Google Scholar] [CrossRef]
- Ren, L.; Wang, Y.; Zhong, J.; Li, X.; Xiao, Y.; Li, J.; Yang, J.; Fan, G.; Guo, L.; Shen, Z. Dynamics of the Upper Respiratory Tract Microbiota and Its Association with Mortality in COVID-19. Am. J. Respir. Crit. Care Med. 2021, 204, 1379–1390. [Google Scholar] [CrossRef]
- Soffritti, I.; D’Accolti, M.; Fabbri, C.; Passaro, A.; Manfredini, R.; Zuliani, G.; Libanore, M.; Franchi, M.; Contini, C.; Caselli, E. Oral Microbiome Dysbiosis Is Associated with Symptoms Severity and Local Immune/Inflammatory Response in COVID-19 Patients: A Cross-Sectional Study. Front. Microbiol. 2021, 12, 687513. [Google Scholar] [CrossRef]
- Hanada, S.; Pirzadeh, M.; Carver, K.Y.; Deng, J.C. Respiratory Viral Infection-Induced Microbiome Alterations and Secondary Bacterial Pneumonia. Front. Immunol. 2018, 9, 2640. [Google Scholar] [CrossRef]
- Abranches, J.; Zeng, L.; Kajfasz, J.K.; Palmer, S.; Chakraborty, B.; Wen, Z.; Richards, V.P.; Brady, L.J.; Lemos, J.A. Biology of Oral Streptococci. Microbiol. Spectr. 2018, 6, 10–1128. [Google Scholar] [CrossRef]
- Shen, Y.; Yu, F.; Zhang, D.; Zou, Q.; Xie, M.; Chen, X.; Yuan, L.; Lou, B.; Xie, G.; Wang, R. Dynamic Alterations in the Respiratory Tract Microbiota of Patients with COVID-19 and Its Association with Microbiota in the Gut. Adv. Sci. 2022, 9, 2200956. [Google Scholar] [CrossRef]
- Dickson, R.P.; Huffnagle, G.B. The Lung Microbiome: New Principles for Respiratory Bacteriology in Health and Disease. PLoS Pathog. 2015, 11, e1004923. [Google Scholar] [CrossRef]
- Horn, K.J.; Jaberi Vivar, A.C.; Arenas, V.; Andani, S.; Janoff, E.N.; Clark, S.E. Corynebacterium Species Inhibit Streptococcus Pneumoniae Colonization and Infection of the Mouse Airway. Front. Microbiol. 2022, 12, 804935. [Google Scholar] [CrossRef]
- Wolff, L.; Martiny, D.; Deyi, V.Y.M.; Maillart, E.; Clevenbergh, P.; Dauby, N. COVID-19–Associated Fusobacterium Nucleatum Bacteremia, Belgium. Emerg. Infect. Dis. 2021, 27, 975. [Google Scholar] [CrossRef] [PubMed]
- Westblade, L.F.; Simon, M.S.; Satlin, M.J. Bacterial Coinfections in Coronavirus Disease 2019. Trends Microbiol. 2021, 29, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, H.H.; Fissel, J.A.; Fanelli, B.; Bergman, Y.; Gniazdowski, V.; Dadlani, M.; Carroll, K.C.; Colwell, R.R.; Simner, P.J. Metagenomic Next-Generation Sequencing of Nasopharyngeal Specimens Collected from Confirmed and Suspect COVID-19 Patients. MBio 2020, 11, e01969-20. [Google Scholar] [CrossRef]
- Dubourg, G.; Yacouba, A.; Bossi, V.; Raoult, D.; Lagier, J.-C. Profile of the Nasopharyngeal Microbiota Affecting the Clinical Course in COVID-19 Patients. Front. Microbiol. 2022, 13, 871627. [Google Scholar]
- Chen, J.; Liu, X.; Liu, W.; Yang, C.; Jia, R.; Ke, Y.; Guo, J.; Jia, L.; Wang, C.; Chen, Y. Comparison of the Respiratory Tract Microbiome in Hospitalized COVID-19 Patients with Different Disease Severity. J. Med. Virol. 2022, 94, 5284–5293. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Zhang, C.; Lyu, J.; Yan, C.; Cao, R.; Pan, M.; Li, Y. Beware of Pharyngeal Fusobacterium Nucleatum in COVID-19. BMC Microbiol. 2021, 21, 277. [Google Scholar] [CrossRef]
- Budhraja, A.; Basu, A.; Gheware, A.; Abhilash, D.; Rajagopala, S.; Pakala, S.; Sumit, M.; Ray, A.; Subramaniam, A.; Mathur, P. Molecular Signature of Postmortem Lung Tissue from COVID-19 Patients Suggests Distinct Trajectories Driving Mortality. Dis. Model. Mech. 2022, 15, dmm049572. [Google Scholar] [CrossRef]
- Cabras, O.; Turmel, J.-M.; Olive, C.; Bigeard, B.; Lehoux, M.; Pierre-Francois, S.; Guitteaud, K.; Abel, S.; Cuzin, L.; Cabié, A. COVID-19 and Pasteurella Multocida Pulmonary Coinfection: A Case Series. Trop. Med. Infect. Dis. 2022, 7, 429. [Google Scholar] [CrossRef]
- Shilts, M.H.; Rosas-Salazar, C.; Strickland, B.A.; Kimura, K.S.; Asad, M.; Sehanobish, E.; Freeman, M.H.; Wessinger, B.C.; Gupta, V.; Brown, H.M. Severe COVID-19 Is Associated with an Altered Upper Respiratory Tract Microbiome. Front. Cell. Infect. Microbiol. 2022, 11, 1436. [Google Scholar] [CrossRef]
- Li, Y.; Liu, J.; Zhu, Y.; Peng, C.; Dong, Y.; Liu, L.; He, Y.; Lu, G.; Zheng, Y. Alterations of Oral Microbiota in Chinese Children with Viral Encephalitis and/or Viral Meningitis. J. Microbiol. 2022, 60, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; D’Souza, G.; Fakhry, C.; Bigelow, E.O.; Usyk, M.; Burk, R.D.; Zhao, N. Oral Human Papillomavirus Associated With Differences in Oral Microbiota Beta Diversity and Microbiota Abundance. J. Infect. Dis. 2022, 226, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.; Li, Y.; Zhao, J. The Gut Microbiome of Healthy Long-Living People. Aging 2019, 11, 289. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Ostan, R.; Candela, M.; Biagi, E.; Brigidi, P.; Capri, M.; Franceschi, C. Gut Microbiota Changes in the Extreme Decades of Human Life: A Focus on Centenarians. Cell Mol. Life Sci. 2018, 75, 129–148. [Google Scholar] [CrossRef]
- Zaneveld, J.R.; McMinds, R.; Vega Thurber, R. Stress and Stability: Applying the Anna Karenina Principle to Animal Microbiomes. Nat. Microbiol. 2017, 2, 17121. [Google Scholar] [CrossRef]
- Merenstein, C.; Bushman, F.D.; Collman, R.G. Alterations in the Respiratory Tract Microbiome in COVID-19: Current Observations and Potential Significance. Microbiome 2022, 10, 165. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Vidal, C.; Sanjuan, G.; Moreno-García, E.; Puerta-Alcalde, P.; Garcia-Pouton, N.; Chumbita, M.; Fernandez-Pittol, M.; Pitart, C.; Inciarte, A.; Bodro, M. Incidence of Co-Infections and Superinfections in Hospitalized Patients with COVID-19: A Retrospective Cohort Study. Clin. Microbiol. Infect. 2021, 27, 83–88. [Google Scholar] [CrossRef]
- Marino, A.; Campanella, E.; Stracquadanio, S.; Ceccarelli, M.; Zagami, A.; Nunnari, G.; Cacopardo, B. Corynebacterium Striatum Bacteremia during SARS-CoV2 Infection: Case Report, Literature Review, and Clinical Considerations. Infect. Dis. Rep. 2022, 14, 383–390. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Positive Test (PT) | Negative Test (NT) | ||||
|---|---|---|---|---|---|
| 182 (71.1%) | 74 (28.9%) | ||||
| Age group | Men, n (%) | Women, n (%) | Men, n (%) | Women | Total |
| Children (0–14 years) | 5 (2%) | 13 (5.1%) | 12 (4.7%) | 4 (1.6%) | 34 (13.3%) |
| Teenagers (15–24 years) | 14 (5.5%) | 15 (5.9%) | 9 (3.5%) | 12 (11.3%) | 50 (19.5%) |
| Adults (25–64 years) | 47 (18.4%) | 63 (24.6%) | 7 (2.7%) | 25 (9.8%) | 142 (55.5%) |
| Elderly (65+ years) | 13 (5.1%) | 12 (4.7%) | 1 (0.4%) | 4 (1.6%) | 30 (11.7%) |
| Total | 79 (30.9%) | 103 (40.2%) | 29 (11.3%) | 45 (17.6%) | 256 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourumeau, W.; Tremblay, K.; Jourdan, G.; Girard, C.; Laprise, C. Bacterial Biomarkers of the Oropharyngeal and Oral Cavity during SARS-CoV-2 Infection. Microorganisms 2023, 11, 2703. https://doi.org/10.3390/microorganisms11112703
Bourumeau W, Tremblay K, Jourdan G, Girard C, Laprise C. Bacterial Biomarkers of the Oropharyngeal and Oral Cavity during SARS-CoV-2 Infection. Microorganisms. 2023; 11(11):2703. https://doi.org/10.3390/microorganisms11112703
Chicago/Turabian StyleBourumeau, William, Karine Tremblay, Guillaume Jourdan, Catherine Girard, and Catherine Laprise. 2023. "Bacterial Biomarkers of the Oropharyngeal and Oral Cavity during SARS-CoV-2 Infection" Microorganisms 11, no. 11: 2703. https://doi.org/10.3390/microorganisms11112703
APA StyleBourumeau, W., Tremblay, K., Jourdan, G., Girard, C., & Laprise, C. (2023). Bacterial Biomarkers of the Oropharyngeal and Oral Cavity during SARS-CoV-2 Infection. Microorganisms, 11(11), 2703. https://doi.org/10.3390/microorganisms11112703