
Unraveling Intestinal Microbial Shifts in ESRD and Kidney Transplantation: Implications for Disease-Related Dysbiosis

Abstract
:1. Introduction
2. Materials and Methods
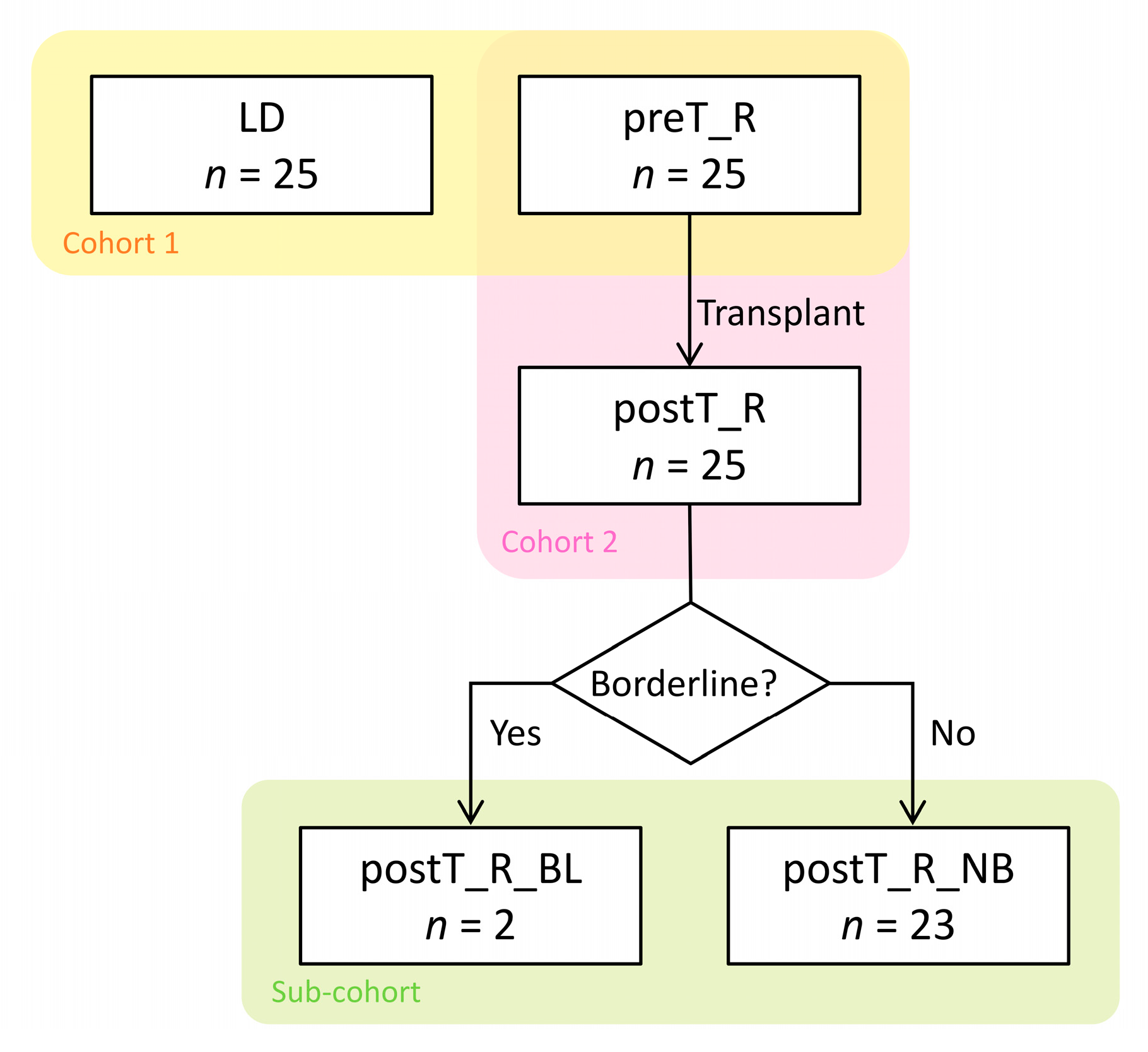
2.1. Transplant Cohort
2.2. Sub-Cohort Selection
2.3. Fecal Specimen Collection and DNA Extraction
2.4. 16S rRNA Deep Sequencing
2.5. Bioinformatics Analysis
2.6. Statistical Analysis
3. Results
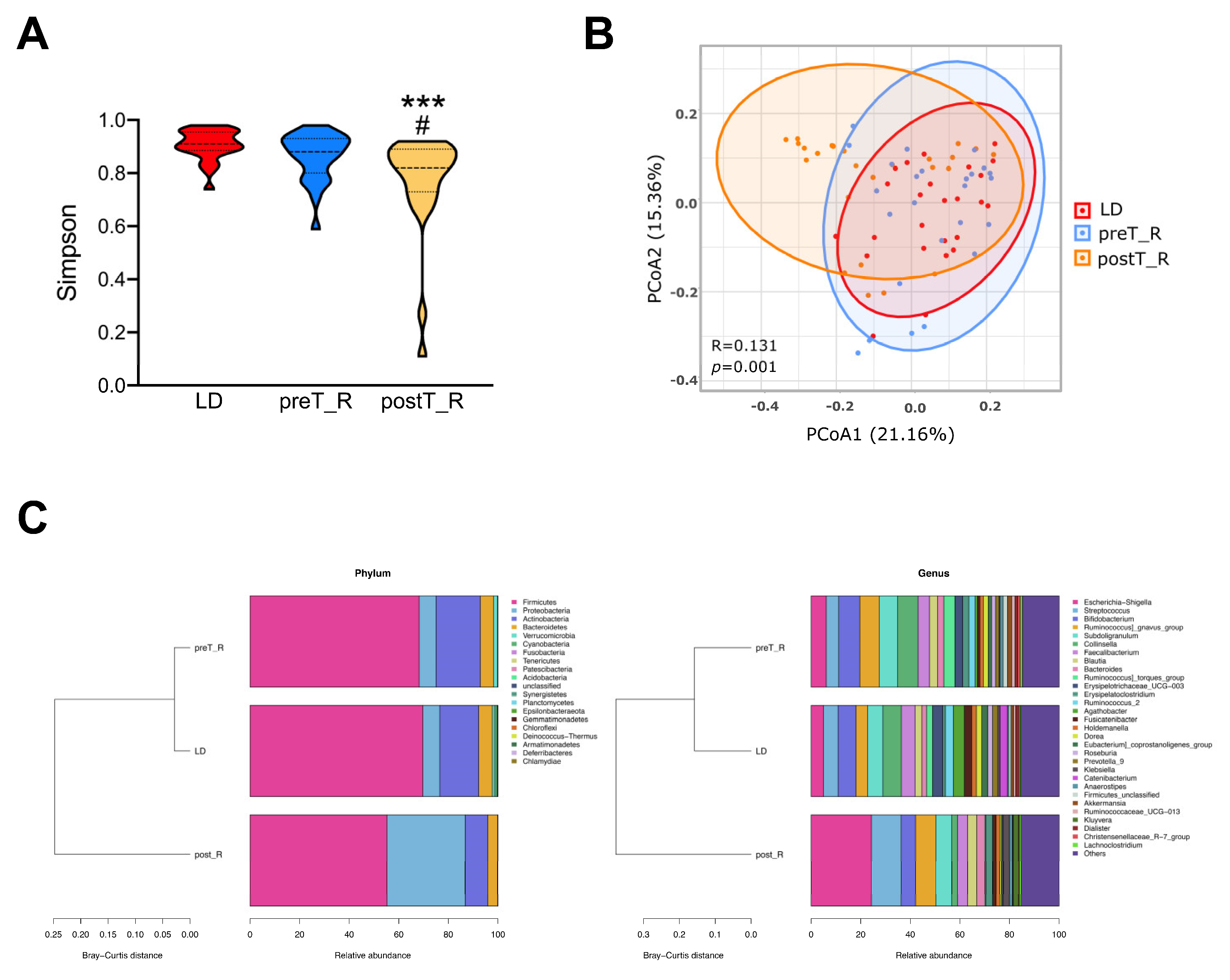
3.1. Similar Microbial Abundance within the LD and ESRD Fecal Specimens
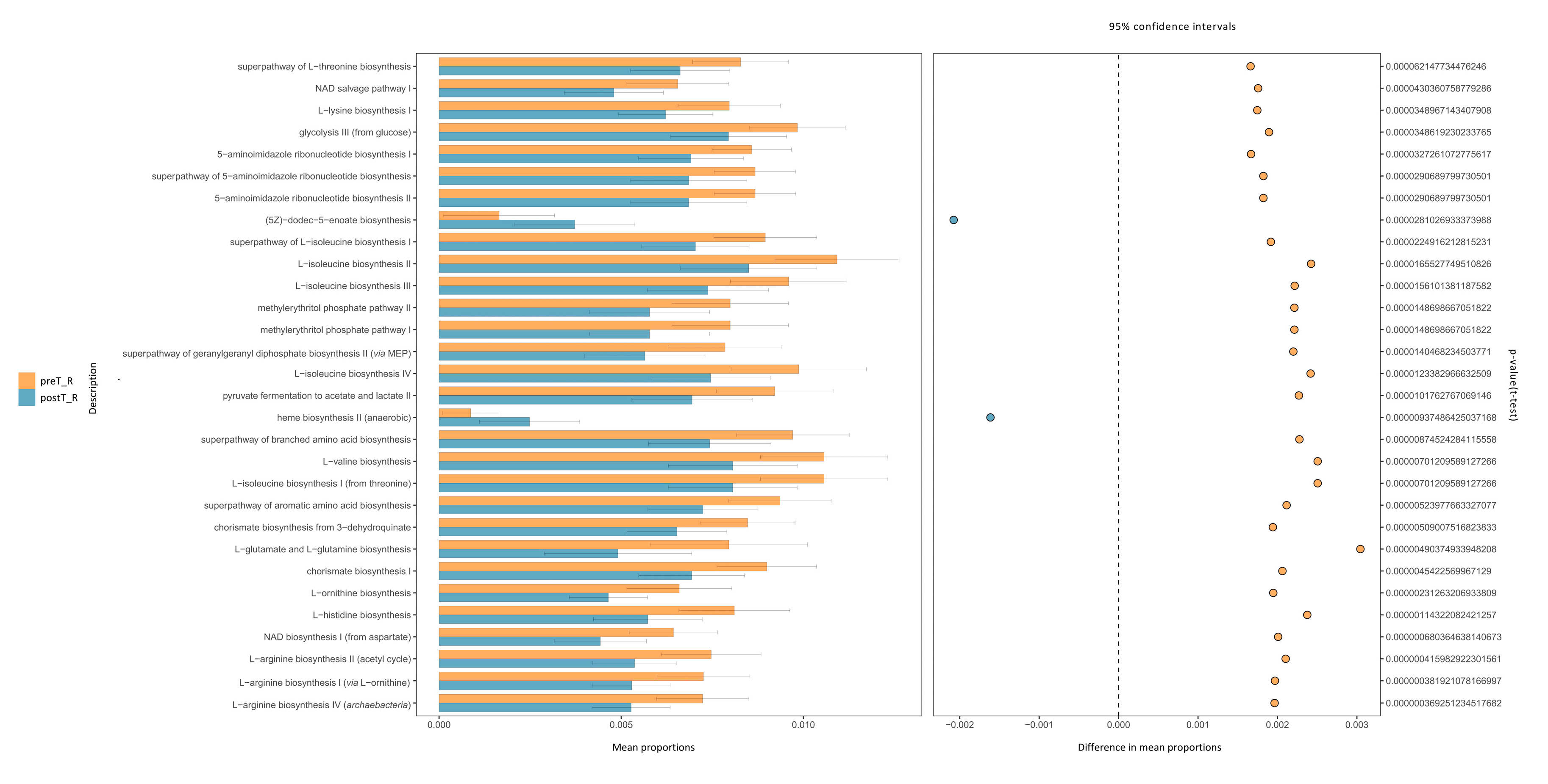
3.2. Changes in the Microbial Population Associated with Kidney Transplant
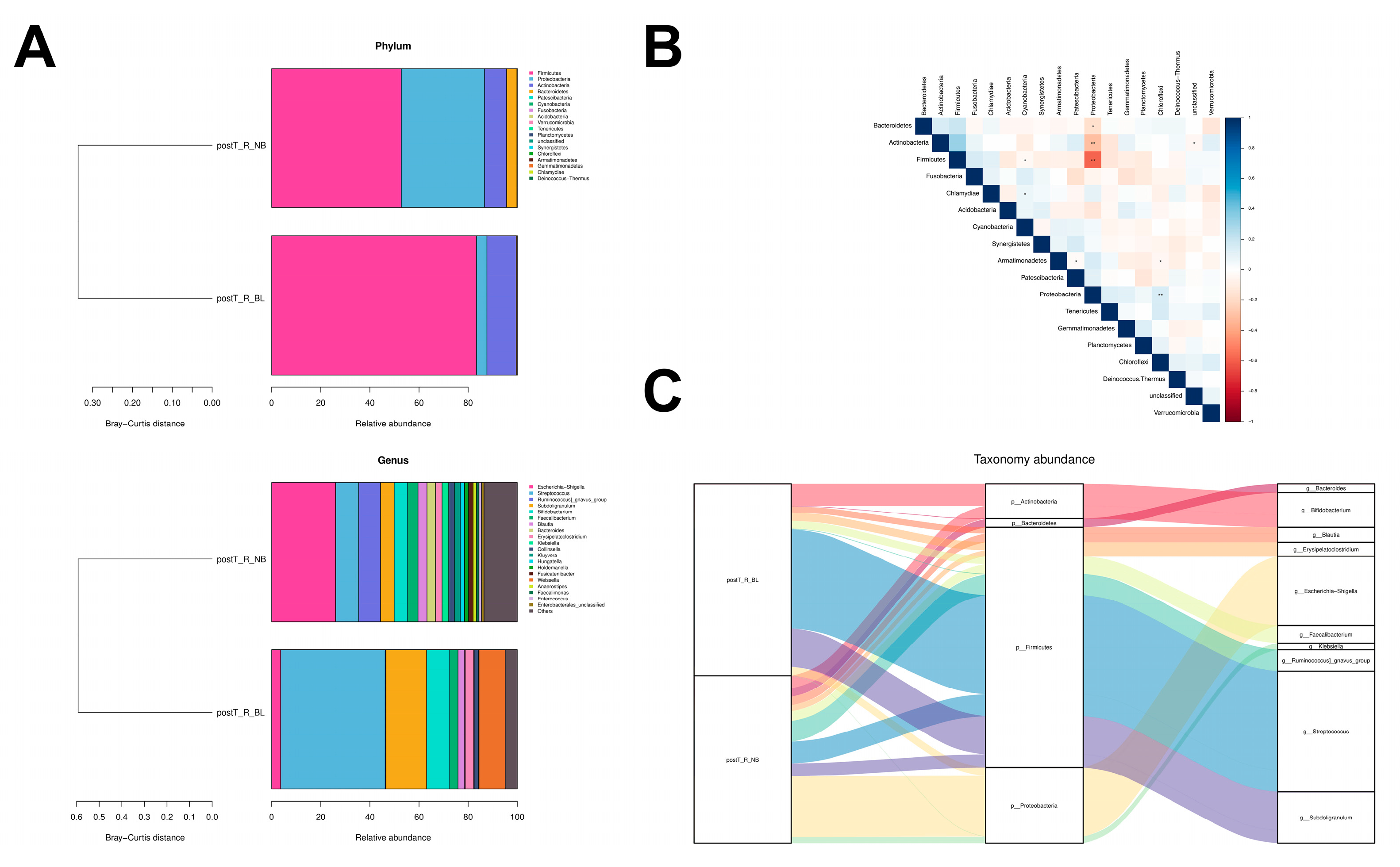
3.3. Bacterial Taxa Associated with Early Rejection Episodes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sugimoto, Y.; Camacho, F.R.; Wang, S.; Chankhamjon, P.; Odabas, A.; Biswas, A.; Jeffrey, P.D.; Donia, M.S. A metagenomic strategy for harnessing the chemical repertoire of the human microbiome. Science 2019, 366, eaax9176. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Theriot, C.M.; Petri, W.A., Jr. Role of Microbiota-Derived Bile Acids in Enteric Infections. Cell 2020, 181, 1452–1454. [Google Scholar] [CrossRef] [PubMed]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef]
- Stein-Thoeringer, C.K.; Nichols, K.B.; Lazrak, A.; Docampo, M.D.; Slingerland, A.E.; Slingerland, J.B.; Clurman, A.G.; Armijo, G.; Gomes, A.L.C.; Shono, Y.; et al. Lactose drives Enterococcus expansion to promote graft-versus-host disease. Science 2019, 366, 1143–1149. [Google Scholar] [CrossRef]
- Shono, Y.; van den Brink, M.R.M. Gut microbiota injury in allogeneic haematopoietic stem cell transplantation. Nat. Rev. Cancer 2018, 18, 283–295. [Google Scholar] [CrossRef]
- Swarte, J.C.; Douwes, R.M.; Hu, S.; Vich Vila, A.; Eisenga, M.F.; van Londen, M.; Gomes-Neto, A.W.; Weersma, R.K.; Harmsen, H.J.M.; Bakker, S.J.L. Characteristics and Dysbiosis of the Gut Microbiome in Renal Transplant Recipients. J. Clin. Med. 2020, 9, 386. [Google Scholar] [CrossRef]
- Wu, H.; Singer, J.; Kwan, T.K.; Loh, Y.W.; Wang, C.; Tan, J.; Li, Y.J.; Lai, S.W.C.; Macia, L.; Alexander, S.I.; et al. Gut Microbial Metabolites Induce Donor-Specific Tolerance of Kidney Allografts through Induction of T Regulatory Cells by Short-Chain Fatty Acids. J. Am. Soc. Nephrol. 2020, 31, 1445–1461. [Google Scholar] [CrossRef]
- Mathewson, N.D.; Jenq, R.; Mathew, A.V.; Koenigsknecht, M.; Hanash, A.; Toubai, T.; Oravecz-Wilson, K.; Wu, S.R.; Sun, Y.; Rossi, C.; et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat. Immunol. 2016, 17, 505–513. [Google Scholar] [CrossRef]
- Fricke, W.F.; Maddox, C.; Song, Y.; Bromberg, J.S. Human microbiota characterization in the course of renal transplantation. Am. J. Transplant. 2014, 14, 416–427. [Google Scholar] [CrossRef]
- Lee, J.R.; Muthukumar, T.; Dadhania, D.; Toussaint, N.C.; Ling, L.; Pamer, E.; Suthanthiran, M. Gut microbial community structure and complications after kidney transplantation: A pilot study. Transplantation 2014, 98, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhang, X.; Chen, F. A cross-sectional study on gut microbiota in patients with chronic kidney disease undergoing kidney transplant or hemodialysis. Am. J. Transl. Res. 2023, 15, 1756–1765. [Google Scholar] [PubMed]
- Pantazi, A.C.; Kassim, M.A.K.; Nori, W.; Tuta, L.A.; Mihai, C.M.; Chisnoiu, T.; Balasa, A.L.; Mihai, L.; Lupu, A.; Frecus, C.E.; et al. Clinical Perspectives of Gut Microbiota in Patients with Chronic Kidney Disease and End-Stage Kidney Disease: Where Do We Stand? Biomedicines 2023, 11, 2480. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, X.; Li, M.; Li, R.; Zhao, M. Shifts in Intestinal Metabolic Profile Among Kidney Transplantation Recipients with Antibody-Mediated Rejection. Ther. Clin. Risk Manag. 2023, 19, 207–217. [Google Scholar] [CrossRef]
- Carron, C.; Pais de Barros, J.P.; Gaiffe, E.; Deckert, V.; Adda-Rezig, H.; Roubiou, C.; Laheurte, C.; Masson, D.; Simula-Faivre, D.; Louvat, P.; et al. End-Stage Renal Disease-Associated Gut Bacterial Translocation: Evolution and Impact on Chronic Inflammation and Acute Rejection After Renal Transplantation. Front. Immunol. 2019, 10, 1630. [Google Scholar] [CrossRef]
- Lee, J.R.; Huang, J.; Magruder, M.; Zhang, L.T.; Gong, C.; Sholi, A.N.; Albakry, S.; Edusei, E.; Muthukumar, T.; Lubetzky, M.; et al. Butyrate-producing gut bacteria and viral infections in kidney transplant recipients: A pilot study. Transpl. Infect. Dis. 2019, 21, e13180. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.R.; Magruder, M.; Zhang, L.; Westblade, L.F.; Satlin, M.J.; Robertson, A.; Edusei, E.; Crawford, C.; Ling, L.; Taur, Y.; et al. Gut microbiota dysbiosis and diarrhea in kidney transplant recipients. Am. J. Transplant. 2019, 19, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Loupy, A.; Haas, M.; Roufosse, C.; Naesens, M.; Adam, B.; Afrouzian, M.; Akalin, E.; Alachkar, N.; Bagnasco, S.; Becker, J.U.; et al. The Banff 2019 Kidney Meeting Report (I): Updates on and clarification of criteria for T cell- and antibody-mediated rejection. Am. J. Transplant. 2020, 20, 2318–2331. [Google Scholar] [CrossRef]
- Logue, J.B.; Stedmon, C.A.; Kellerman, A.M.; Nielsen, N.J.; Andersson, A.F.; Laudon, H.; Lindstrom, E.S.; Kritzberg, E.S. Experimental insights into the importance of aquatic bacterial community composition to the degradation of dissolved organic matter. ISME J. 2016, 10, 533–545. [Google Scholar] [CrossRef]
- Yang, T.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat. Rev. Nephrol. 2018, 14, 442–456. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Jiang, H.; Shi, K.; Ren, Y.; Zhang, P.; Cheng, S. Gut bacterial translocation is associated with microinflammation in end-stage renal disease patients. Nephrology 2012, 17, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Kamada, N.; Chen, G.Y.; Inohara, N.; Nunez, G. Control of pathogens and pathobionts by the gut microbiota. Nat. Immunol. 2013, 14, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Bian, B.; Teng, L.; Nelson, C.D.; Driver, J.; Elzo, M.A.; Jeong, K.C. Host genetic effects upon the early gut microbiota in a bovine model with graduated spectrum of genetic variation. ISME J. 2020, 14, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef]
- Taur, Y.; Xavier, J.B.; Lipuma, L.; Ubeda, C.; Goldberg, J.; Gobourne, A.; Lee, Y.J.; Dubin, K.A.; Socci, N.D.; Viale, A.; et al. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin. Infect. Dis. 2012, 55, 905–914. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Mey, A.R.; Gomez-Garzon, C.; Payne, S.M. Iron Transport and Metabolism in Escherichia, Shigella, and Salmonella. EcoSal Plus 2021, 9, eESP00342020. [Google Scholar] [CrossRef]
- Li, X.; Li, R.; Ji, B.; Zhao, L.; Wang, J.; Yan, T. Integrative metagenomic and metabolomic analyses reveal the role of gut microbiota in antibody-mediated renal allograft rejection. J. Transl. Med. 2022, 20, 614. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mean Relative Abundance (%) | BH-Adjusted p-Value | |||
|---|---|---|---|---|
| preT_R | postT_R | Difference | ||
| Phylum level | ||||
| p__Proteobacteria | 6.83 | 31.54 | 24.71 | 0.01 |
| p__Actinobacteria | 17.89 | 9.18 | 8.71 | 0.05 |
| p__Bacteroidetes | 5.26 | 3.92 | 1.34 | 0.05 |
| Genus level | ||||
| g__Escherichia-Shigella | 6.10 | 24.29 | 18.19 | 0.06 |
| g__Streptococcus | 5.02 | 12.02 | 7.01 | 0.07 |
| g__Collinsella | 8.20 | 2.26 | 5.94 | 0.08 |
| g__Ruminococcus_torques_group | 4.46 | 0.44 | 4.01 | 0.02 |
| g__Ruminococcus_2 | 2.57 | 0.01 | 2.56 | 0.12 |
| g__Klebsiella | 0.09 | 2.44 | 2.35 | 0.03 |
| g__Firmicutes_unclassified | 1.81 | 0.09 | 1.72 | 0.00 |
| g__Dorea | 1.97 | 0.63 | 1.34 | 0.05 |
| g__Roseburia | 1.63 | 0.36 | 1.26 | 0.14 |
| g__Ruminococcaceae_UCG-013 | 1.40 | 0.20 | 1.20 | 0.04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, P.; Luo, S.; Guo, L.; Wang, X.; Ren, X.; Lv, J.; Chen, Y.; Lin, X.; Chen, J.; Wang, R. Unraveling Intestinal Microbial Shifts in ESRD and Kidney Transplantation: Implications for Disease-Related Dysbiosis. Microorganisms 2023, 11, 2747. https://doi.org/10.3390/microorganisms11112747
Yan P, Luo S, Guo L, Wang X, Ren X, Lv J, Chen Y, Lin X, Chen J, Wang R. Unraveling Intestinal Microbial Shifts in ESRD and Kidney Transplantation: Implications for Disease-Related Dysbiosis. Microorganisms. 2023; 11(11):2747. https://doi.org/10.3390/microorganisms11112747
Chicago/Turabian StyleYan, Pengpeng, Sulin Luo, Luying Guo, Xingxia Wang, Xue Ren, Junhao Lv, Ying Chen, Xinyu Lin, Jianghua Chen, and Rending Wang. 2023. "Unraveling Intestinal Microbial Shifts in ESRD and Kidney Transplantation: Implications for Disease-Related Dysbiosis" Microorganisms 11, no. 11: 2747. https://doi.org/10.3390/microorganisms11112747