
Predominance of Recombinant Norovirus Strains in Greece, 2016–2018
 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Stool Specimens and Viral RNA Extraction
2.2. Genotypic Characterization of Norovirus Strainsand Phylogenetic Analysis
2.3. Detection and Analysis of Recombinant Strains
3. Results
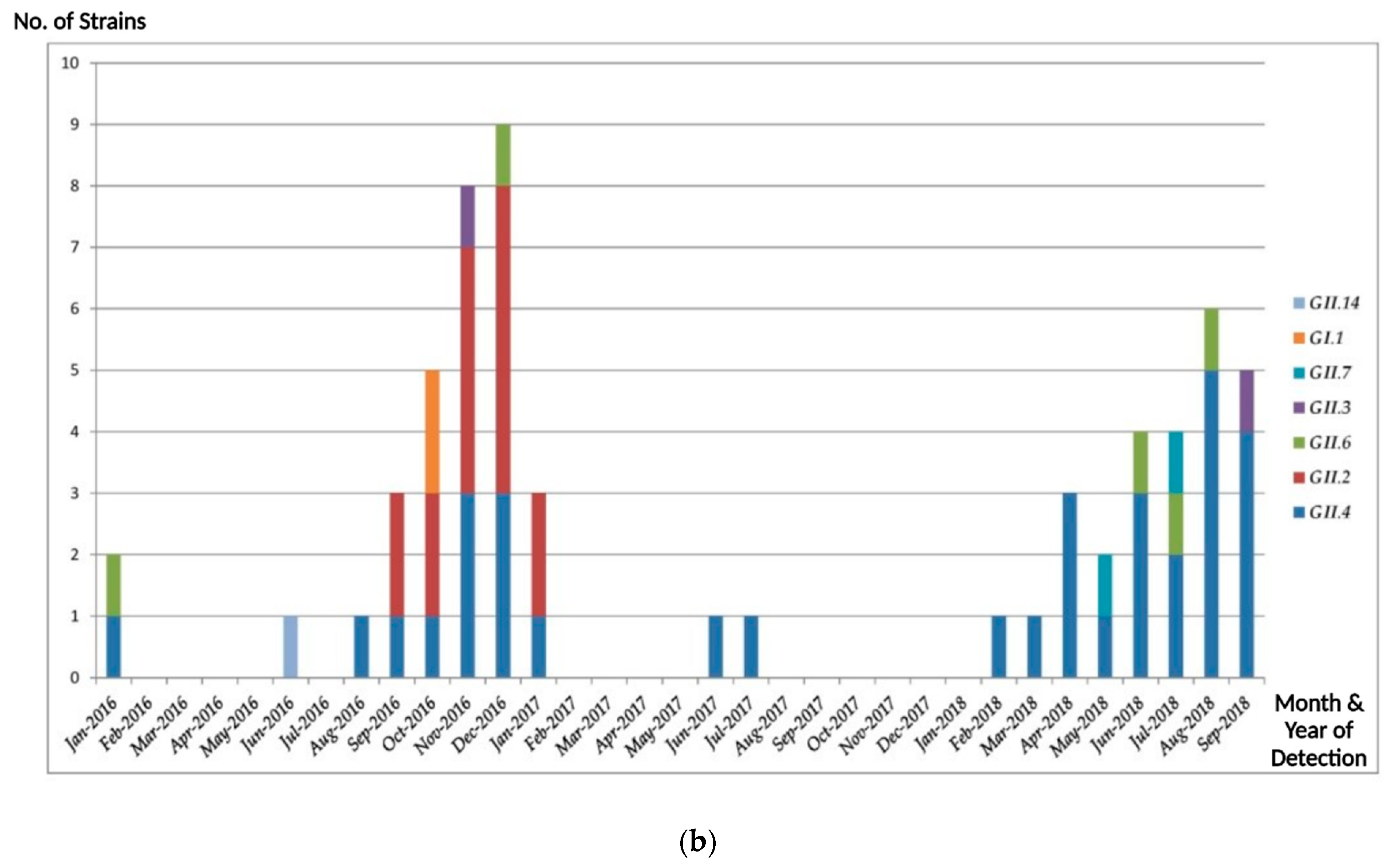
3.1. Norovirus Genotype Distribution
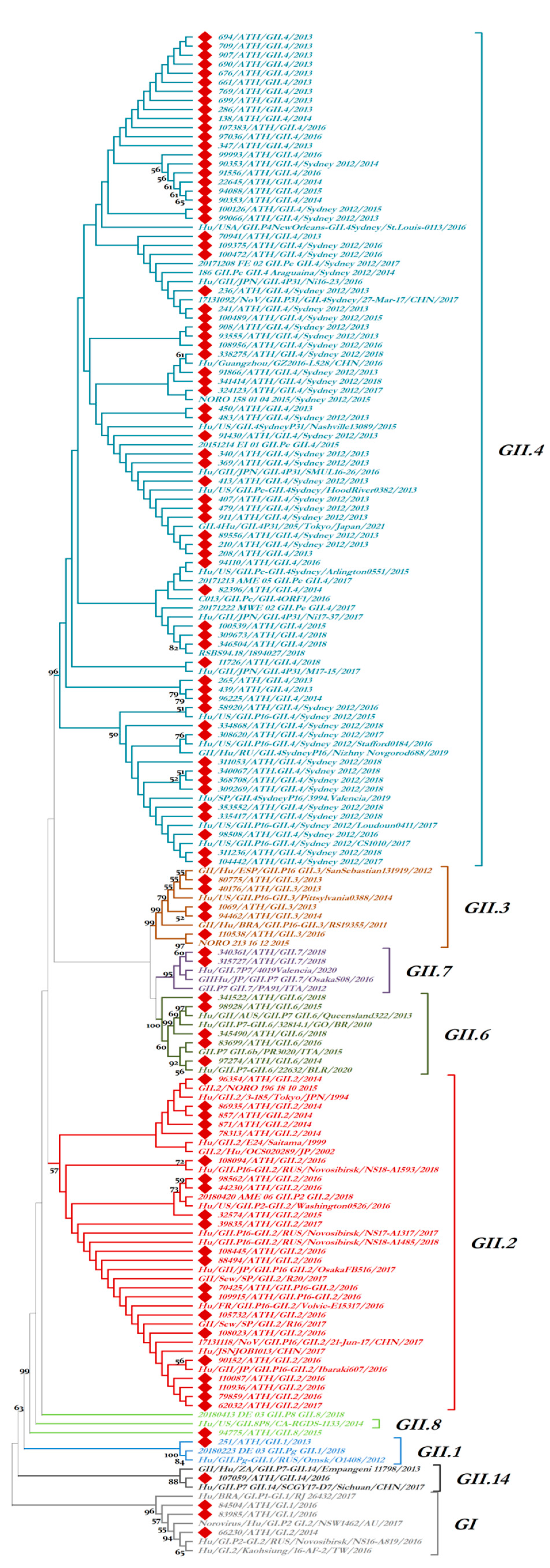
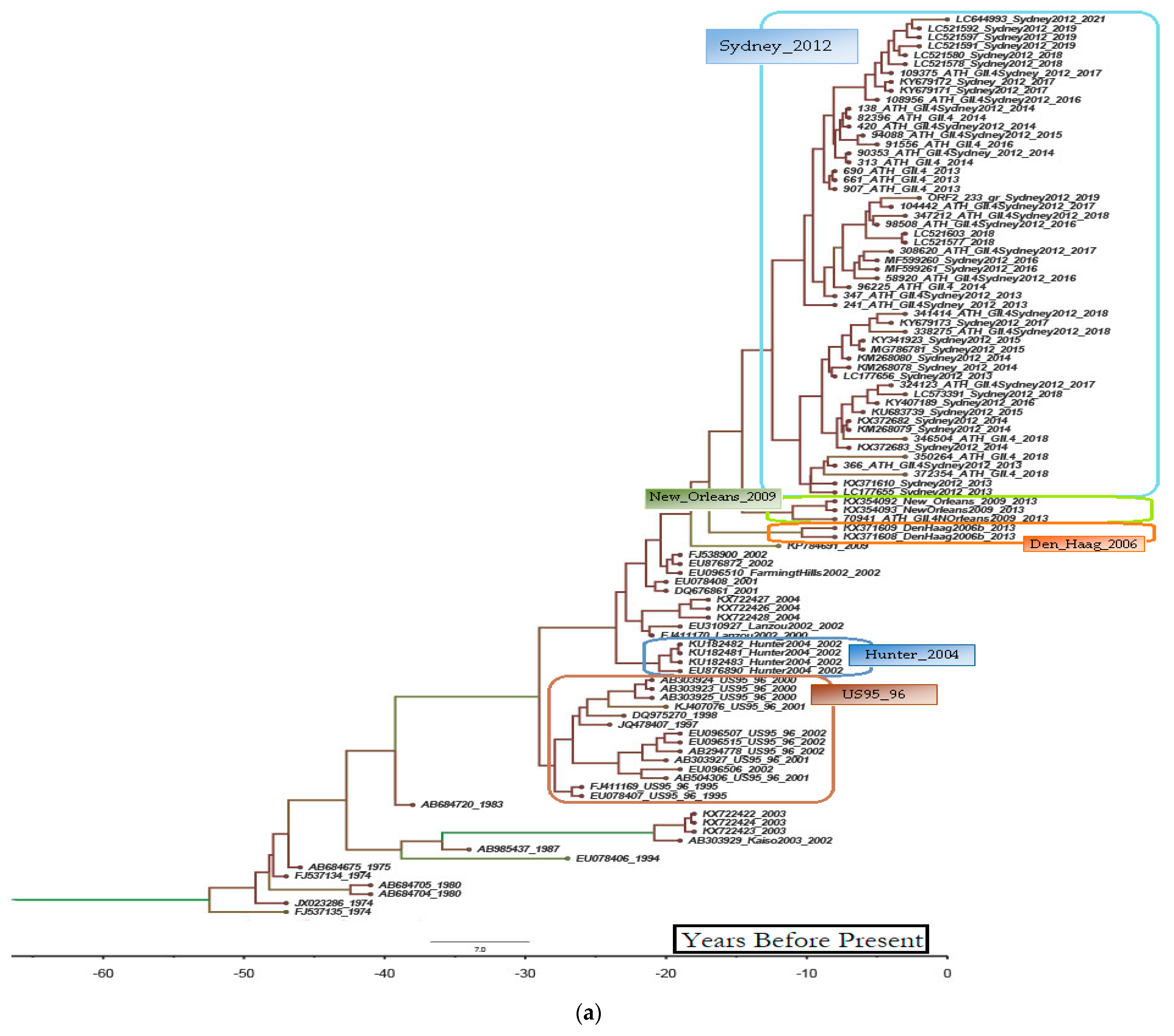
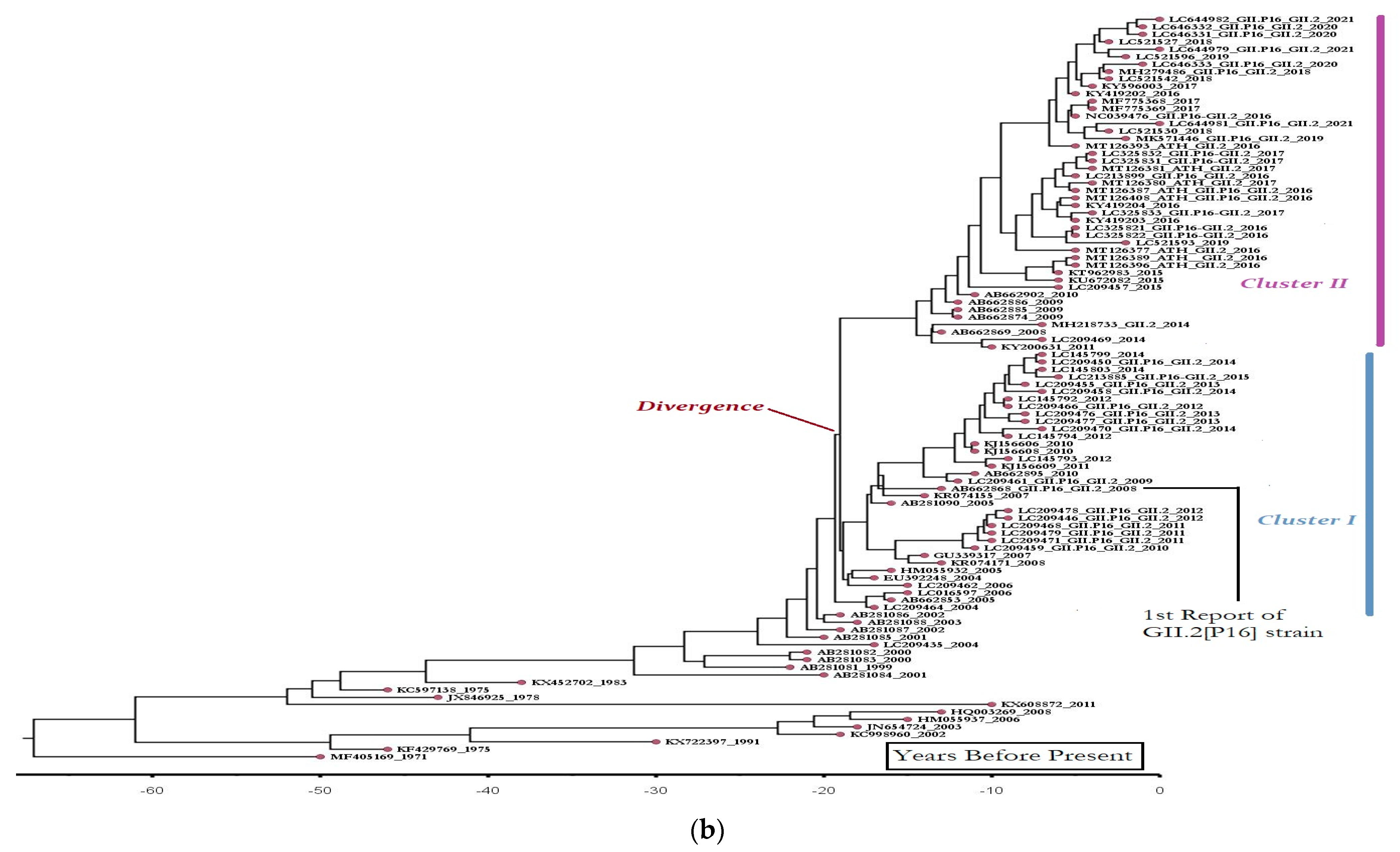
3.2. Epidemiologic and Phylogenetic Analysis
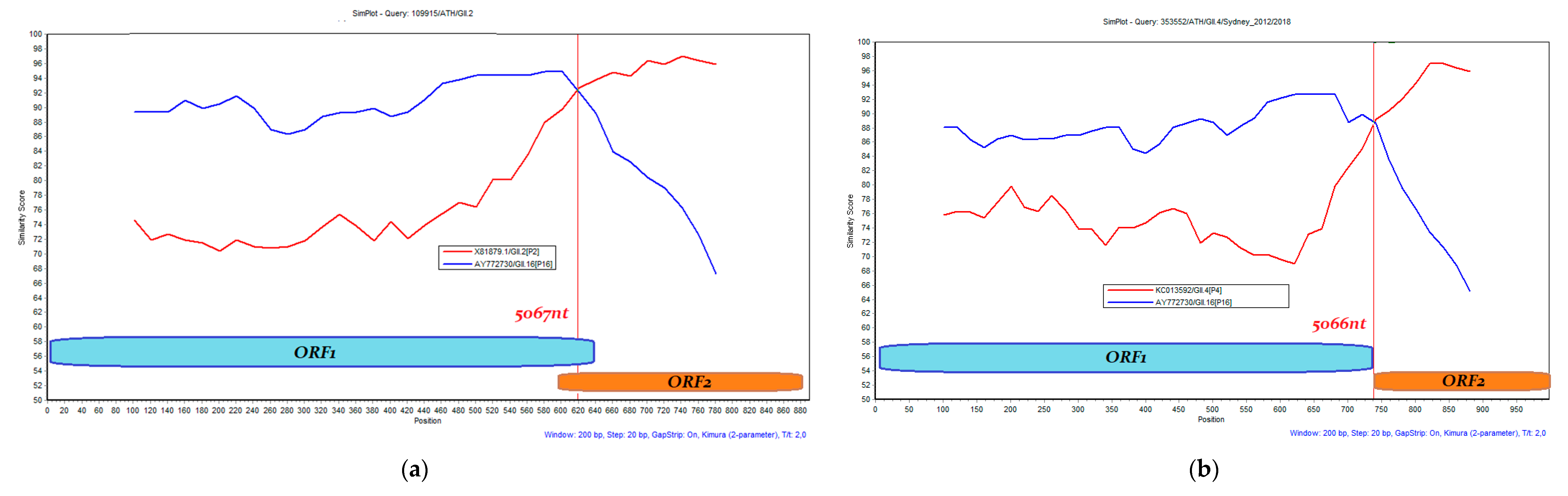
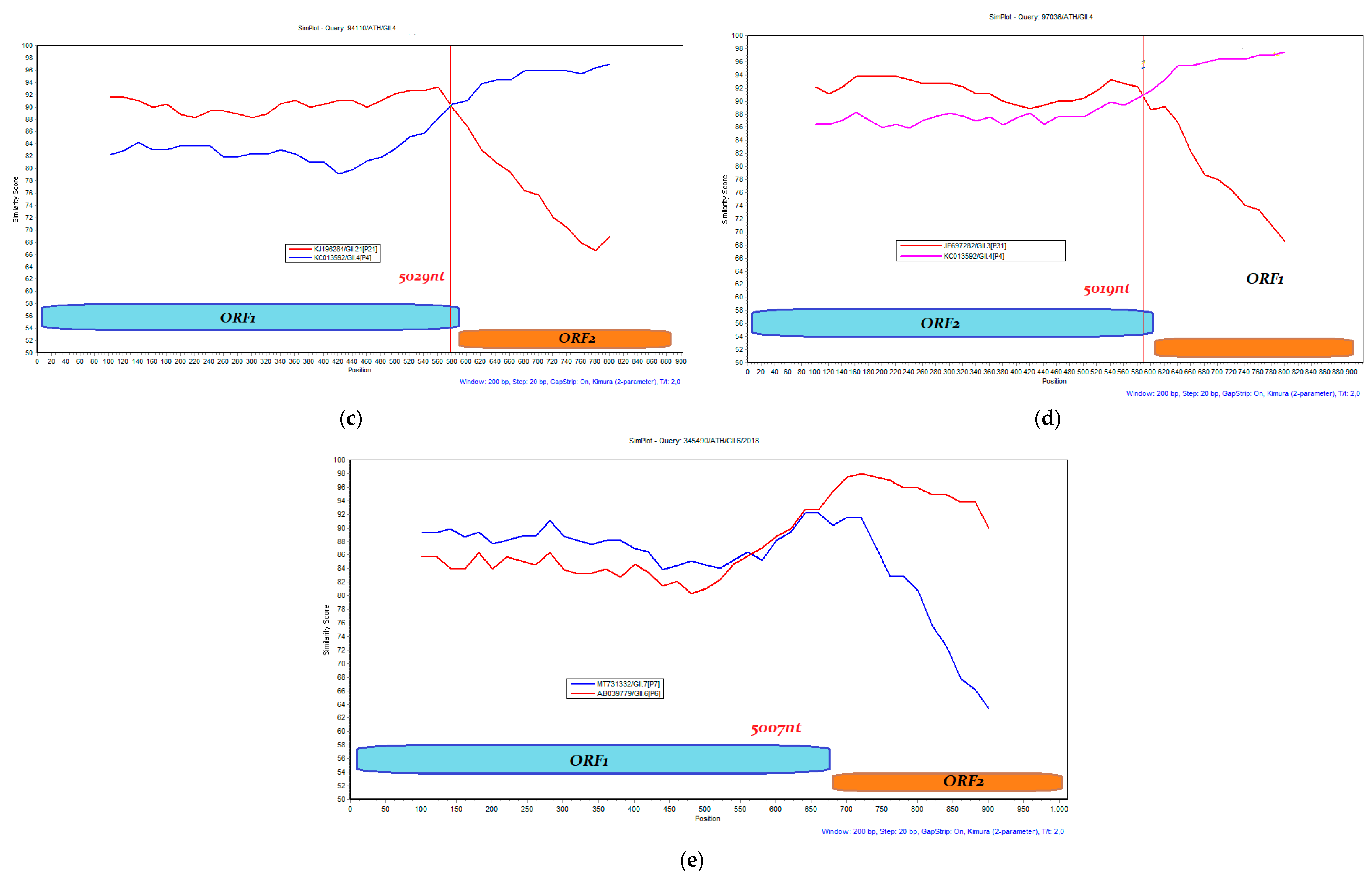
3.3. Recombination Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Robilotti, E.; Deresinski, S.; Pinsky, B.A. Norovirus. Clin. Microbiol. Rev. 2015, 28, 134–164. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, L.; Hong, X.; Gao, J.; Zuo, Y.; Liang, Y.; Jiang, Y.; Zhang, J.; Wu, A.; Xue, L.; et al. The VP2 protein exhibits cross-interaction to the VP1 protein in norovirus GII.17. Infect. Genet. Evol. 2022, 100, 105265. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, P.; de Graaf, M.; Parra, G.I.; Chan, M.C.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H.; et al. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2019, 100, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.J. Noroviruses: The perfect human pathogens? J. Infect. Dis. 2012, 205, 1622–1624. [Google Scholar] [CrossRef] [PubMed]
- Roger, I.; Glass, R.I.; Umesh, D.; Parashar, U.D.; Estes, M.D. Norovirus gastroenteritis. N. Engl. J. Med. 2009, 361, 1776–1785. [Google Scholar]
- Patel, M.M.; Widdowson, M.A.; Glass, R.I.; Akazawa, K.; Vinjé, J.; Parashar, U.D. Systematic literature review of role of noroviruses in sporadic gastroenteritis. Emerg. Infect. Dis. 2008, 14, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, T.K.; Desai, R.; Hall, A.J.; Patel, M.; Parashar, U.D.; Lopman, B.A. Clinical characteristics of norovirus-associated deaths: A systematic literature review. Am. J. Infect. Control 2013, 41, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.I. Emergence of norovirus strains: A tale of two genes. Virus Evol. 2019, 5, vez048. [Google Scholar] [CrossRef]
- Vega, E.; Barclay, L.; Gregoricus, N.; Shirley, S.H.; Lee, D.; Vinje, J. Genotypic and epidemiologic trends of norovirus outbreaks in the United States, 2009 to 2013. J. Clin. Microbiol. 2014, 52, 147–155. [Google Scholar] [CrossRef]
- Kumazaki, M.; Usuku, S. Genetic analysis of norovirus GII.4 variant strains detected in outbreaks of gastroenteritis in Yokohama, Japan, from the 2006–2007 to the 2013–2014 seasons. PLoS ONE 2015, 10, e0142568. [Google Scholar] [CrossRef]
- Tohma, K.; Lepore, C.J.; Gao, Y.; Ford-Siltz, L.A.; Parra, G.I. Population Genomics of GII.4 Noroviruses Reveal Complex Diversification and New Antigenic Sites Involved in the Emergence of Pandemic Strains. mBio 2019, 10, e02202-19. [Google Scholar] [CrossRef]
- Gao, Z.; Liu, B.; Huo, D.; Yan, H.; Jia, L.; Du, Y.; Qian, H.; Yang, Y.; Wang, X.; Li, J.; et al. Increased norovirus activity was associated with a novel norovirus GII.17 variant in Beijing, China during winter 2014–2015. BMC Infect. Dis. 2015, 15, 574. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.L.; Zhu, Z.X.; Cui, J.L.; Yu, J.M. Evolutionary analyses of emerging GII.2[P16] and GII.4 Sydney [P16] noroviruses. Virus Evol. 2022, 8, veac030. [Google Scholar] [CrossRef] [PubMed]
- Siafakas, N.; Zerva, L.; Hatzaki, D.; Lebessi, E.; Chronopoulou, G.; Paraskakis, I.; Pournaras, S. Molecular epidemiology of noroviruses in children in South Greece, 2013–2015. J. Med. Virol. 2018, 90, 1703–1711. [Google Scholar] [CrossRef] [PubMed]
- Kojima, S.; Kageyama, T.; Fukushi, S.; Hoshino, F.B.; Shinohara, M.; Uchida, K.; Natori, K.; Takeda, N.; Katayama, K. Genogroup-specific PCR primers for detection of Norwalk-like viruses. J. Virol. Methods 2002, 100, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Vinjé, J.; Hamidjaja, R.A.; Sobsey, M.D. Development and application of a capsid VP1 (region D) based reverse transcription PCR assay for genotyping of genogroup I and II noroviruses. J. Virol. Methods 2004, 116, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Kroneman, A.; Vennema, H.; Deforchem, K.; van der Avoort, H.; Peñaranda, S.; Oberste, M.S.; Vinjé, J.; Koopmans, M. An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. 2011, 51, 121–125. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Vinjé, J.; Koopmans, M.P. Molecular detection and epidemiology of small round-structured viruses in outbreaks of gastroenteritis in the Netherlands. J. Infect. Dis. 1996, 174, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Samson, S.; Lord, É.; Makarenkov, V. SimPlot++: A Python application for representing sequence similarity and detecting recombination. Bioinformatics 2022, 38, 3118–3120. [Google Scholar] [CrossRef] [PubMed]
- Oldak, E.; Sulik, A.; Rozkiewicz, D.; Liwoch-Nienartowicz, N. Norovirus infections in children under 5 years of age hospitalized due to the acute viral gastroenteritis in northeastern Poland. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 417–422. [Google Scholar] [CrossRef]
- Zeng, M.; Xu, X.; Zhu, C.; Lin, S.; Jie, Y.; Shu, X. Chinese pediatric study group of norovirus. Clinical and molecular epidemiology of norovirus infection in childhood diarrhoea in China. J. Med. Virol. 2012, 84, 145–151. [Google Scholar] [CrossRef]
- Tan, D.; Deng, L.; Wang, M.; Li, X.; Ma, Y.; Liu, W. High prevalence and genetic diversity of noroviruses among children with sporadic acute gastroenteritis in Nanning City, China, 2010–2011. J. Med. Virol. 2015, 87, 498–503. [Google Scholar] [CrossRef]
- Zhou, N.; Zhang, H.; Lin, X.; Hou, P.; Wang, S.; Tao, Z.; Bi, Z.; Xu, A. A waterborne norovirus gastroenteritis outbreak in a school, in Eastern China. Epidemiol. Infect. 2016, 144, 1212–1219. [Google Scholar] [CrossRef]
- Ruether, I.G.; Tsakogiannis, D.; Kyriakopoulou, Z.; Dimitriou, T.G.; Papamichail, C.; Gartzonika, C.; Leveidiotou-Stefanou, S.; Markoulatos, P. Circulation of intergenotype recombinant noroviruses GII.9/GII.6 from 2006 to 2011 in central Greece. Virus Genes 2014, 48, 23–31. [Google Scholar] [CrossRef]
- Tryfinopoulou, K.; Kyritsi, M.; Mellou, K.; Kolokythopoulou, F.; Mouchtouri, V.A.; Potamiti-Komi, M.; Lamprou, A.; Georgakopoulou, T.; Hadjichristodoulou, C. Norovirus waterborne outbreak in Chalkidiki, Greece, 2015: Detection of GI.P2_GI.2 and GII.P16_GII.13 unusual strains. Epidemiol. Infect. 2019, 147, e227. [Google Scholar] [CrossRef]
- Parra, G.I.; Squires, R.B.; Karangwa, C.K.; Johnson, J.A.; Lepore, C.J.; Sosnovtsev, S.V.; Green, K.Y. Static and Evolving Norovirus Genotypes: Implications for Epidemiology and Immunity. PLoS Pathog. 2017, 13, e1006136. [Google Scholar] [CrossRef]
- Chan, M.C.; Lee, N.; Hung, T.N.; Kwok, K.; Cheung, K.; Tin, E.K.; Lai, R.W.; Nelson, E.A.; Leung, T.F.; Chan, P.K. Rapid emergence and predominance of a broadly recognizing and fast-evolving norovirus GII.17 variant in late 2014. Nat. Commun. 2015, 6, 10061. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, Y.; Ishikawa, M.; Shimizu, T.; Komane, A.; Kasuo, S.; Shinohara, M.; Nagasawa, K.; Kimura, H.; Ryo, A.; Okabe, N.; et al. Genetic analyses of GII.17 norovirus strains in diarrheal disease outbreaks from December 2014 to March 2015 in Japan reveal a novel polymerase sequence and amino acid substitutions in the capsid region. Eurosurveillance 2015, 20, 21173. [Google Scholar] [CrossRef] [PubMed]
- Tohma, K.; Lepore, C.J.; Ford-Siltz, L.A.; Parra, G.I. Phylogenetic Analyses Suggest that Factors Other Than the Capsid Protein Play a Role in the Epidemic Potential of GII.2 Norovirus. mSphere 2017, 2, e00187-17. [Google Scholar] [CrossRef] [PubMed]
- Khamrin, P.; Kumthip, K.; Yodmeeklin, A.; Jampanil, N.; Phengma, P.; Yamsakul, P.; Okitsu, S.; Kobayashi, T.; Ushijima, H.; Maneekarn, N. Changing Predominance of Norovirus Recombinant Strains GII.2[P16] to GII.4[P16] and GII.4[P31] in Thailand, 2017 to 2018. Microbiol. Spectr. 2022, 10, e0044822. [Google Scholar] [CrossRef]
- Han, J.; Wu, X.; Chen, L.; Fu, Y.; Xu, D.; Zhang, P.; Ji, L. Emergence of norovirus GII.P16-GII.2 strains in patients with acute gastroenteritis in Huzhou, China, 2016–2017. BMC Infect. Dis. 2018, 18, 342. [Google Scholar] [CrossRef]
- Iritani, N.; Kaida, A.; Abe, N.; Sekiguchi, J.; Kubo, H.; Takakura, K.; Goto, K.; Ogura, H.; Seto, Y. Increase of GII.2 norovirus infections during the 2009–2010 season in Osaka City, Japan. J. Med. Virol. 2012, 84, 517–525. [Google Scholar] [CrossRef]
- Eden, J.S.; Tanaka, M.M.; Boni, M.F.; Rawlinson, W.D.; White, P.A. Recombination within the pandemic norovirus GII.4 lineage. J. Virol. 2013, 87, 6270–6282. [Google Scholar] [CrossRef]
- Ruis, C.; Roy, S.; Brown, J.R.; Allen, D.J.; Goldstein, R.A.; Breuer, J. The emerging GII.P16-GII.4 Sydney 2012 norovirus lineage is circulating worldwide, arose by late-2014 and contains polymerase changes that may increase virus transmission. PLoS ONE 2017, 12, e0179572. [Google Scholar] [CrossRef]
- Ruis, C.; Lindesmith, L.C.; Mallory, M.L.; Brewer-Jensen, P.D.; Bryant, J.M.; Costantini, V.; Monit, C.; Vinjé, J.; Baric, R.S.; Goldstein, R.A.; et al. Preadaptation of pandemic GII.4 noroviruses in unsampled virus reservoirs years before emergence. Virus Evol. 2020, 6, veaa067. [Google Scholar] [CrossRef]
- Kim, M.S.; Koo, E.S.; Choi, Y.S.; Kim, J.Y.; Yoo, C.H.; Yoon, H.J.; Kim, T.O.; Choi, H.B.; Kim, J.H.; Choi, J.D.; et al. Distribution of Human Norovirus in the Coastal Waters of South Korea. PLoS ONE 2016, 11, e0163800. [Google Scholar] [CrossRef]
- van Beek, J.; de Graaf, M.; Al-Hello, H.; Allen, D.J.; Ambert-Balay, K.; Botteldoorn, N.; Brytting, M.; Buesa, J.; Cabrerizo, M.; Chan, M.; et al. Molecular surveillance of norovirus, 2005–2016: An epidemiological analysis of data collected from the NoroNet network. Lancet Infect. Dis. 2018, 18, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.C.; Roy, S.; Bonifacio, J.; Zhang, L.Y.; Chhabra, P.; Chan, J.C.M.; Celma, C.; Igoy, M.A.; Lau, S.L.; Mohammad, K.N.; et al. Detection of Norovirus Variant GII.4 Hong Kong in Asia and Europe, 2017–2019. Emerg. Infect. Dis. 2021, 27, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.A.; Eden, J.S.; Rawlinson, W.D.; White, P.A. Rapid evolution of pandemic noroviruses of the GII.4 lineage. PLoS Pathog. 2010, 26, e1000831. [Google Scholar] [CrossRef]
- Arias, A.; Thorne, L.; Ghurburrun, E.; Bailey, D.; Goodfellow, I. Norovirus Polymerase Fidelity Contributes to Viral Transmission In Vivo. mSphere 2016, 19, e00279-16. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GenBank Accession No. | Position on NoV Genome | Isolate | Stool Collection Date | Genotype ORF2 (VP1) | Genotype ORF1 (RdRp) | Recombination Breakpoint Position (nt) | GII.4 Pandemic Variant |
|---|---|---|---|---|---|---|---|
| MT126392.1 | 5047–5309 | 83699/ATH/GII.6 | Jan-2016 | GII.6 | Not Available | - | - |
| MT126388.1 | 5049–5349 | 109375/ATH/GII.4 | Jan-2016 | GII.4 | Not Available | - | Sydney_2012 |
| MT126407.1 | 5047–5344 | 107059/ATH/GII.14 | Jun-2016 | GII.14 | Not Available | - | - |
| MT126405.1 | 5047–5343 | 58920/ATH/GII.4 | Aug-2016 | GII.4 | Not Available | - | Sydney_2012 |
| MT126384.2 | 5047–5326 | 90152/ATH/GII.2 | Sep-2016 | GII.2 | Not Available | - | - |
| MT126402.1 | 5047–5339 | 108956/ATH/GII.4 | Sep-2016 | GII.4 | Not Available | - | Sydney_2012 |
| MT126389.1 | 5047–5337 | 98562/ATH/GII.2 | Sep-2016 | GII.2 | Not Available | - | - |
| MT126387.1 | 4327–5351 | 109915/ATH/GII.2 | Oct-2016 | GII.2 | GII.P16 | 5067 (MG746035 Ref.) | - |
| MT126393.1 | 5047–5353 | 108094/ATH/GII.2 | Oct-2016 | GII.2 | Not Available | - | - |
| MT126398.1 | 5047–5344 | 100472/ATH/GII.4 | Oct-2016 | GII.4 | Not Available | - | Sydney_2012 |
| MT129796.1 | 5326–5627 | 84504/ATH/GI.1 | Oct-2016 | GI.1 | Not Available | - | - |
| MT129795.1 | 5326–5609 | 83985/ATH/GI.1 | Oct-2016 | GI.1 | Not Available | - | - |
| MT126395.1 | 5047–5344 | 105732/ATH/GII.2 | Nov-2016 | GII.2 | Not Available | - | - |
| MT126397.2 | 4446–5349 | 94110/ATH/GII.4 | Nov-2016 | GII.4 | GII.P21 (GII.Pb) | 5029 (MH218671 Ref.) | Sydney_2012 |
| MT126396.2 | 4326–5337 | 44230/ATH/GII.2 | Nov-2016 | GII.2 | GII.P2 | - | - |
| MT126408.1 | 4836–5356 | 110087/ATH/GII.2 | Nov-2016 | GII.2 | GII.P16 | 5067 (MG746035 Ref.) | - |
| MT126401.1 | 4506–5328 | 107383/ATH/GII.4 | Nov-2016 | GII.4 | GII.P31 (GII.Pe) | 5019 (MK789435 Ref.) | Sydney_2012 |
| MT126391.1 | 4323–5339 | 98508/ATH/GII.4 | Nov-2016 | GII.4 | GII.P16 | 5066 (MK753032 Ref.) | Sydney_2012 |
| MT126385.1 | 5047–5338 | 110538/ATH/GII.3 | Nov-2016 | GII.3 | Not Available | - | - |
| MT126379.1 | 4384–5337 | 88494/ATH/GII.2 | Nov-2016 | GII.2 | GII.P16 | 5067 (MG746035 Ref.) | - |
| MT126400.1 | 4337–5349 | 97036/ATH/GII.4 | Dec-2016 | GII.4 | GII.P31 (GII.Pe) | 5019 (MK789435 Ref.) | Sydney_2012 |
| MT126399.1 | 5047–5349 | 99993/ATH/GII.4 | Dec-2016 | GII.4 | Not Available | - | Sydney_2012 |
| MT126404.1 | 5047–5349 | 91556/ATH/GII.4 | Dec-2016 | GII.4 | Not Available | - | Sydney_2012 |
| MT126390.1 | 5063–5352 | 103242/ATH/GII.6 | Dec-2016 | GII.6 | Not Available | - | - |
| MT126394.2 | 5047–5344 | 110936/ATH/GII.2 | Dec-2016 | GII.2 | Not Available | - | - |
| MT126378.1 | 4447–5349 | 108445/ATH/GII.2 | Dec-2016 | GII.2 | GII.P16 | 5067 (MG746035 Ref.) | - |
| MT126377.1 | 5047–5337 | 79859/ATH/GII.2 | Dec-2016 | GII.2 | Not Available | - | - |
| MT126406.1 | 5047–5322 | 108023/ATH/GII.2 | Dec-2016 | GII.2 | Not Available | - | - |
| MT126386.1 | 4797–5312 | 70425/ATH/GII.2 | Dec-2016 | GII.2 | GII.P16 | 5067 (MG746035 Ref.) | - |
| MT126382.1 | 5047–5329 | 104442/ATH/GII.4 | Jan-2017 | GII.4 | Not Available | - | Sydney_2012 |
| MT126381.1 | 5047–5337 | 39835/ATH/GII.2 | Jan-2017 | GII.2 | Not Available | - | - |
| MT126380.1 | 5047–5328 | 62032/ATH/GII.2 | Jan-2017 | GII.2 | Not Available | - | - |
| MT126383.1 | 5047–5349 | 308620/ATH/GII.4 | Jun-2017 | GII.4 | Not Available | - | Sydney_2012 |
| MT126403.1 | 5047–5328 | 324123/ATH/GII.4 | Jul-2017 | GII.4 | Not Available | - | Sydney_2012 |
| OP557585.1 | 5047–5325 | 11726/ATH/GII.4/2018 | Feb-2018 | GII.4 | Not Available | - | Could not assign |
| OP557581.1 | 4425–5189 | 306792/ATH/GII.4/Sydney_2012/2018 | Mar-2018 | GII.4 | GII.P16 | 5066 (MK753032 Ref.) | Sydney_2012 |
| OP557589.1 | 5044–5349 | 309673/ATH/GII.4/2018 | Apr-2018 | GII.4 | Not Available | - | Could not assign |
| OP557580.1 | 4333–5371 | 311236/ATH/GII.4/Sydney_2012/2018 | Apr-2018 | GII.4 | GII.P16 | 5066 (MK753032 Ref.) | Sydney_2012 |
| OP557587.1 | 4335–5348 | 309269/ATH/GII.4/Sydney_2012/2018 | Apr-2018 | GII.4 | GII.P16 | 5066 (MK753032 Ref.) | Sydney_2012 |
| OP557584.1 | 5048–5327 | 311053/ATH/GII.4/Sydney_2012/2018 | May-2018 | GII.4 | Not Available | - | Sydney_2012 |
| OP557579.1 | 5048–5299 | 315727/ATH/GII.7/2018 | May-2018 | GII.7 | Not Available | - | - |
| OP557592.1 | 4346–5232 | 332648/ATH/GII.6/2018 | Jun-2018 | GII.6 | GII.P7 | 5007 (MW661284 Ref.) | - |
| OP557591.1 | 5047–5356 | 335417/ATH/GII.4/Sydney_2012/2018 | Jun-2018 | GII.4 | Not Available | - | Sydney_2012 |
| OP557590.1 | 5047–5356 | 334868/ATH/GII.4/Sydney_2012/2018 | Jun-2018 | GII.4 | Not Available | - | Sydney_2012 |
| OP557572.1 | 4329–5362 | 338275/ATH/GII.4/Sydney_2012/2018 | Jun-2018 | GII.4 | GII.P31 (GII.Pe) | 5019 (MK789435 Ref.) | Sydney_2012 |
| OP557593.1 | 4506–5331 | 341522/ATH/GII.6/2018 | Jul-2018 | GII.6 | GII.P7 | 5007 (MW661284 Ref.) | - |
| OP557582.1 | 4376–5308 | 340067/ATH.GII.4/Sydney_2012/2018 | Jul-2018 | GII.4 | GII.P16 | 5066 (MK753032 Ref.) | Sydney_2012 |
| OP557575.1 | 4433–5363 | 341414/ATH/GII.4/Sydney_2012/2018 | Jul-2018 | GII.4 | GII.P31 (GII.Pe) | 5019 (MK789435 Ref.) | Sydney_2012 |
| OP557574.1 | 5047–5313 | 340361/ATH/GII.7/2018 | Jul-2018 | GII.7 | Not Available | - | - |
| OP557594.1 | 5066–5309 | 347211/ATH/GII.4/Sydney_2012/2018 | Aug-2018 | GII.4 | Not Available | - | Sydney_2012 |
| OP557586.1 | 4314–5366 | 353552/ATH/GII.4/Sydney_2012/2018 | Aug-2018 | GII.4 | GII.P16 | 5066 (MK753032 Ref.) | Sydney_2012 |
| OP557583.1 | 4353–5364 | 345490/ATH/GII.6/2018 | Aug-2018 | GII.6 | GII.P7 | 5007 (MW661284 Ref.) | - |
| OP557571.1 | 4390–5363 | 346504/ATH/GII.4/2018 | Aug-2018 | GII.4 | GII.P31 (GII.Pe) | 5014 (KC175323 Ref.) | Could not assign |
| OP557570.1 | 4436–5298 | 347212/GII.4/Sydney_2012/2018 | Aug-2018 | GII.4 | GII.P16 | 5066 (MK753032 Ref.) | Sydney_2012 |
| OP557569.1 | 5048–5304 | 350264/ATH/GII.4/2018 | Aug-2018 | GII.4 | Not Available | - | Could not assign |
| OP557578.1 | 5048–5307 | 372354/ATH/GII.4/2018 | Sep-2018 | GII.4 | Not Available | - | Could not assign |
| OP557577.1 | 4323–5266 | 356299/ATH/GII.4/Sydney_2012/2018 | Sep-2018 | GII.4 | GII.P16 | 5066 (MK753032 Ref.) | Sydney_2012 |
| OP557576.1 | 5105–5326 | 372382/ATH/GII.3/2018 | Sep-2018 | GII.3 | Not Available | - | - |
| OP557588.1 | 4326–5324 | 368708/ATH/GII.4/Sydney_2012/2018 | Sep-2018 | GII.4 | GII.P16 | 5066 (MK753032 Ref.) | Sydney_2012 |
| OP557573.1 | 4340–5221 | 358253/ATH/GII.4/Sydney_2012/2018 | Sep-2018 | GII.4 | GII.P16 | 5066 (MK753032 Ref.) | Sydney_2012 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siafakas, N.; Anastassopoulou, C.; Lafazani, M.; Chronopoulou, G.; Rizos, E.; Pournaras, S.; Tsakris, A. Predominance of Recombinant Norovirus Strains in Greece, 2016–2018. Microorganisms 2023, 11, 2885. https://doi.org/10.3390/microorganisms11122885
Siafakas N, Anastassopoulou C, Lafazani M, Chronopoulou G, Rizos E, Pournaras S, Tsakris A. Predominance of Recombinant Norovirus Strains in Greece, 2016–2018. Microorganisms. 2023; 11(12):2885. https://doi.org/10.3390/microorganisms11122885
Chicago/Turabian StyleSiafakas, Nikolaos, Cleo Anastassopoulou, Maria Lafazani, Genovefa Chronopoulou, Emmanouil Rizos, Spyridon Pournaras, and Athanasios Tsakris. 2023. "Predominance of Recombinant Norovirus Strains in Greece, 2016–2018" Microorganisms 11, no. 12: 2885. https://doi.org/10.3390/microorganisms11122885