

Multi-Generation Ecosystem Selection of Rhizosphere Microbial Communities Associated with Plant Genotype and Biomass in Arabidopsis thaliana

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Multi-Generation Selection of Soil Ecosystem
2.2. DNA Extraction and 16S rRNA Amplicon Library Prep
2.3. Sequence Data Analysis and Statistics
2.4. Diversity Analyses
2.5. Adonis
2.6. Neutral Model
2.7. Differential Abundance
2.8. Spearman Correlation for Differentially Abundant OTUs
3. Results
3.1. Changes in Above-Ground Plant Biomass
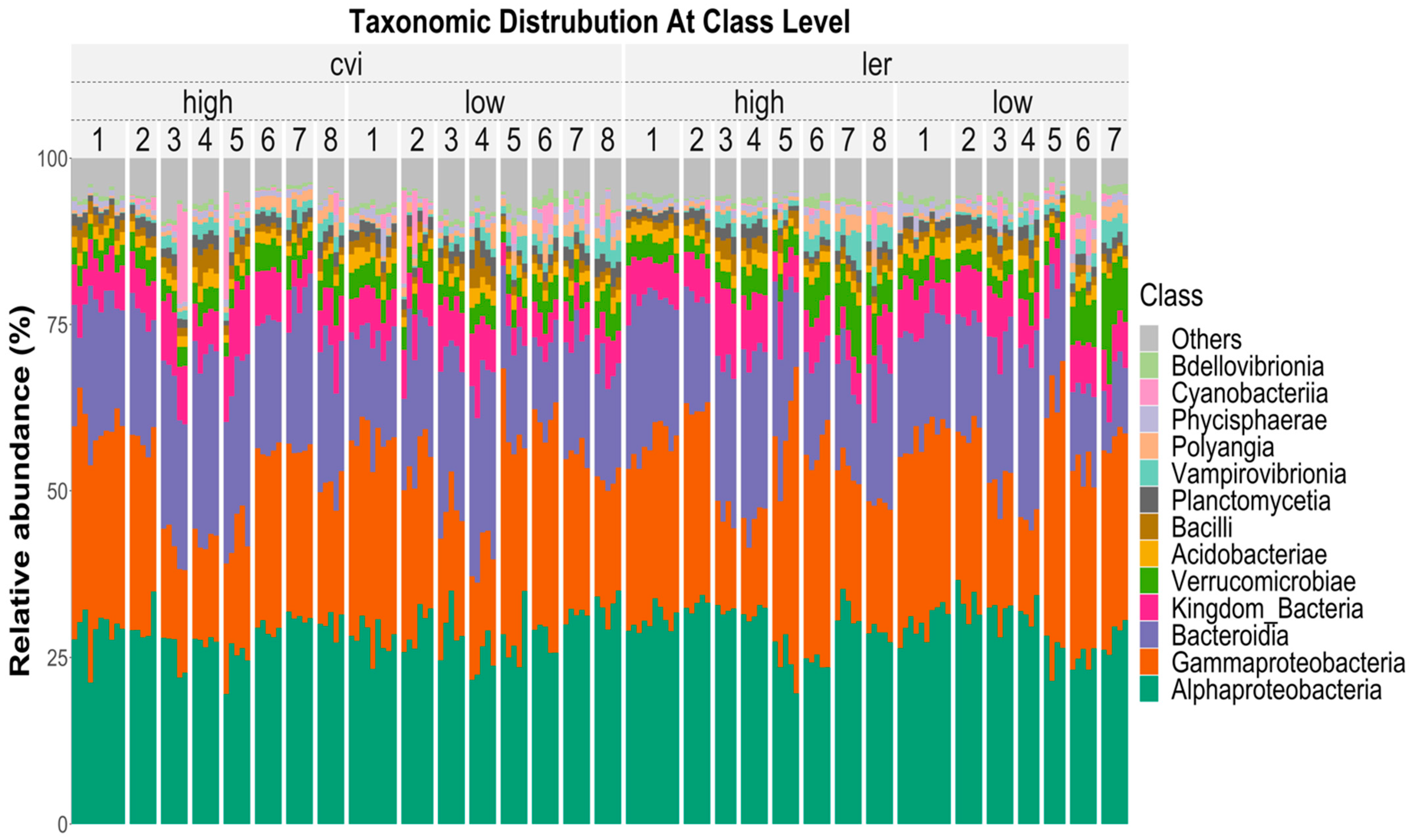
3.2. Microbial Community Composition
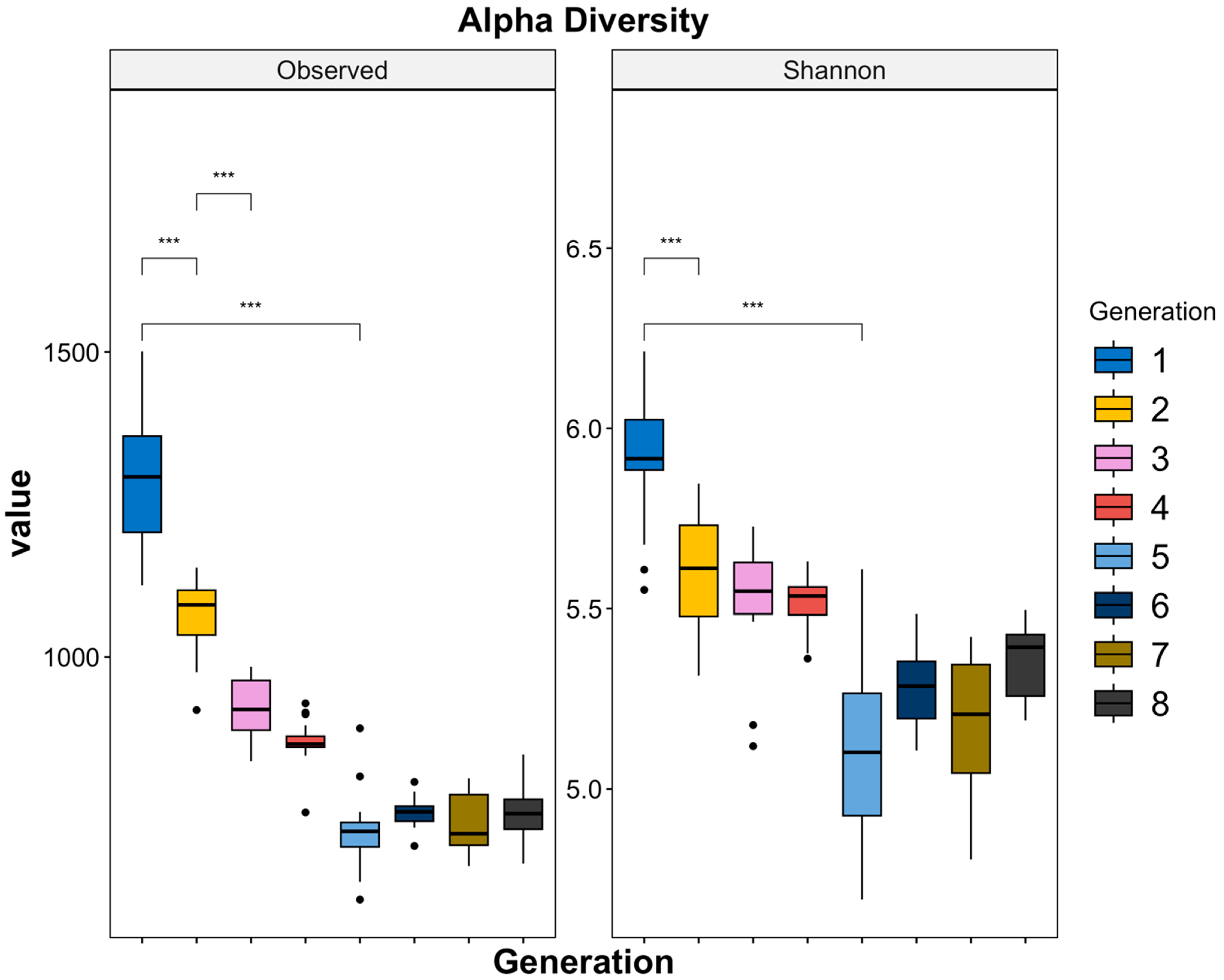
3.3. Trends in Alpha Diversity
3.4. Beta Diversity and Principal Coordinate Analysis
3.5. Sloan Neutral Model
3.6. Differential Abundance and Correlation Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gouda, S.; Kerry, R.G.; Das, G.; Paramithiotis, S.; Shin, H.S.; Patra, J.K. Revitalization of plant growth promoting rhizobacteria for sustainable development in agriculture. Microbiol. Res. 2018, 206, 131–140. [Google Scholar] [CrossRef]
- Jones, D.L.; Nguyen, C.; Finlay, R.D. Carbon flow in the rhizosphere: Carbon trading at the soil-root interface. Plant Soil 2009, 321, 5–33. [Google Scholar] [CrossRef]
- Dennis, P.G.; Miller, A.J.; Hirsch, P.R. Are root exudates more important than other sources of rhizodeposits in structuring rhizosphere bacterial communities? FEMS Microbiol. Ecol. 2010, 72, 313–327. [Google Scholar] [CrossRef]
- Hartmann, A.; Schmid, M.; van Tuinen, D.; Berg, G. Plant-driven selection of microbes. Plant Soil 2009, 321, 235–257. [Google Scholar] [CrossRef]
- Chaparro, J.M.; Sheflin, A.M.; Manter, D.K.; Vivanco, J.M. Manipulating the soil microbiome to increase soil health and plant fertility. Biol. Fertil. Soils 2012, 48, 489–499. [Google Scholar] [CrossRef]
- Marschner, P.; Crowley, D.; Yang, C.H. Development of specific rhizosphere bacterial communities in relation to plant species, nutrition and soil type. Plant Soil 2004, 261, 199–208. [Google Scholar] [CrossRef]
- Garbeva, P.; van Veen, J.A.; van Elsas, J.D. Microbial diversity in soil: Selection microbial populations by plant and soil type and implications for disease suppressiveness. Annu. Rev. Phytopathol. 2004, 42, 243–270. [Google Scholar] [CrossRef] [PubMed]
- Tkacz, A.; Cheema, J.; Chandra, G.; Grant, A.; Poole, P.S. Stability and succession of the rhizosphere microbiota depends upon plant type and soil composition. ISME J. 2015, 9, 2349–2359. [Google Scholar] [CrossRef]
- Berg, G.; Smalla, K. Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol. Ecol. 2009, 68, 1–13. [Google Scholar] [CrossRef]
- Jin, H.; Yang, X.; Liu, R.; Yan, Z.; Li, X.; Li, X.; Su, A.; Zhao, Y.; Qin, B. Bacterial community structure associated with the rhizosphere soils and roots of Stellera chamaejasme L. along a Tibetan elevation gradient. Ann. Microbiol. 2018, 68, 273–286. [Google Scholar] [CrossRef]
- Barbosa Lima, A.; Cannavan, F.S.; Navarrete, A.A.; Teixeira, W.G.; Kuramae, E.E.; Tsai, S.M. Amazonian Dark Earth and Plant Species from the Amazon Region Contribute to Shape Rhizosphere Bacterial Communities. Microb. Ecol. 2015, 69, 855–866. [Google Scholar] [CrossRef]
- Marques, J.M.; da Silva, T.F.; Vollu, R.E.; Blank, A.F.; Ding, G.C.; Seldin, L.; Smalla, K. Plant age and genotype affect the bacterial community composition in the tuber rhizosphere of field-grown sweet potato plants. FEMS Microbiol. Ecol. 2014, 88, 424–435. [Google Scholar] [CrossRef]
- Bodenhausen, N.; Bortfeld-Miller, M.; Ackermann, M.; Vorholt, J.A. A Synthetic Community Approach Reveals Plant Genotypes Affecting the Phyllosphere Microbiota. PLoS Genet. 2014, 10, e1004283. [Google Scholar] [CrossRef]
- Zancarini, A.; Mougel, C.; Voisin, A.S.; Prudent, M.; Salon, C.; Munier-Jolain, N. Soil Nitrogen Availability and Plant Genotype Modify the Nutrition Strategies of M. truncatula and the Associated Rhizosphere Microbial Communities. PLoS ONE 2012, 7, e47096. [Google Scholar] [CrossRef]
- Wei, S.; Liu, B.; Ni, K.; Ma, L.; Shi, Y.; Leng, Y.; Zheng, S.; Gao, S.; Yang, X.; Ruan, J. Rhizosphere Microbial Community Shows a Greater Response Than Soil Properties to Tea (Camellia sinensis L.) Cultivars. Agronomy 2023, 13, 221. [Google Scholar] [CrossRef]
- Yuan, J.; Chaparro, J.M.; Manter, D.K.; Zhang, R.; Vivanco, J.M.; Shen, Q. Roots from distinct plant developmental stages are capable of rapidly selecting their own microbiome without the influence of environmental and soil edaphic factors. Soil Biol. Biochem. 2015, 89, 206–209. [Google Scholar] [CrossRef]
- Houlden, A.; Timms-Wilson, T.M.; Day, M.J.; Bailey, M.J. Influence of plant developmental stage on microbial community structure and activity in the rhizosphere of three field crops. FEMS Microbiol. Ecol. 2008, 65, 193–201. [Google Scholar] [CrossRef]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Rio, T.G.D.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2013, 488, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; Ver, E.; Themaat, L.V.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Coleman-Derr, D.; Desgarennes, D.; Fonseca-Garcia, C.; Gross, S.; Clingenpeel, S.; Woyke, T.; North, G.; Visel, A.; Partida-Martinez, L.P.; Tringe, S.G. Plant compartment and biogeography affect microbiome composition in cultivated and native Agave species. New Phytol. 2016, 209, 798–811. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef]
- Wagner, M.R.; Lundberg, D.S.; Del Rio, T.G.; Tringe, S.G.; Dangl, J.L.; Mitchell-Olds, T. Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat Commun. 2016, 7, 12151. [Google Scholar] [CrossRef] [PubMed]
- Schlaeppi, K.; Dombrowski, N.; Oter, R.G.; Ver Loren Van Themaat, E.; Schulze-Lefert, P. Quantitative divergence of the bacterial root microbiota in Arabidopsis thaliana relatives. Proc. Natl. Acad. Sci. USA 2014, 111, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Robert, C.A.M.; Cadot, S.; Zhang, X.; Ye, M.; Li, B.; Manzo, D.; Chervet, N.; Steinger, T.; Van Der Heijden, M.G.; et al. Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nat. Commun. 2018, 9, 2738. [Google Scholar] [CrossRef] [PubMed]
- Van der Putten, W.H.; Bardgett, R.D.; Bever, J.D.; Bezemer, T.M.; Casper, B.B.; Fukami, T.; Kardol, P.; Klironomos, J.N.; Kulmatiski, A.; Schweitzer, J.A.; et al. Plant-soil feedbacks: The past, the present and future challenges. J. Ecol. 2013, 101, 265–276. [Google Scholar] [CrossRef]
- Panke-buisse, K.; Poole, A.C.; Goodrich, J.K.; Ley, R.E.; Kao-kniffin, J. Selection on soil microbiomes reveals reproducible impacts on plant function. ISME J. 2014, 9, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Swenson, W.; Wilson, D.S.; Elias, R. Artificial ecosystem selection. Proc. Natl. Acad. Sci. USA 2000, 97, 9110–9114. [Google Scholar] [CrossRef] [PubMed]
- Mueller, U.G.; Juenger, T.E.; Kardish, M.R.; Carlson, A.L.; Burns, K.M.; Edwards, J.A.; Smith, C.C.; Fang, C.C.; Des Marais, D.L. Artificial Selection on Microbiomes To Breed Microbiomes That Confer Salt Tolerance to Plants. mSystems 2021, 6, e01125-21. [Google Scholar] [CrossRef]
- Lau, J.A.; Lennon, J.T. Rapid responses of soil microorganisms improve plant fitness in novel environments. Proc. Natl. Acad. Sci. USA 2012, 109, 14058–14062. [Google Scholar] [CrossRef]
- terHorst, C.P.; Lennon, J.T.; Lau, J.A. The relative importance of rapid evolution for plant-microbe interactions depends on ecological context. Proc. R. Soc. B 2014, 281, 20140028. [Google Scholar] [CrossRef]
- Jochum, M.D.; McWilliams, K.L.; Pierson, E.A.; Jo, Y.K. Host-mediated microbiome engineering (HMME) of drought tolerance in the wheat rhizosphere. PLoS ONE 2019, 14, e0225933. [Google Scholar] [CrossRef]
- Cordovez, V.; Rotoni, C.; Dini-Andreote, F.; Oyserman, B.; Carrión, V.J.; Raaijmakers, J.M. Successive plant growth amplifies genotype-specific assembly of the tomato rhizosphere microbiome. Sci. Total Environ. 2021, 772, 144825. [Google Scholar] [CrossRef] [PubMed]
- Morella, N.M.; Weng, F.C.H.; Joubert, P.M.; Metcalf, C.J.E.; Lindow, S.; Koskella, B. Successive passaging of a plant-associated microbiome reveals robust habitat and host genotype-dependent selection. Proc. Natl. Acad. Sci. USA 2020, 117, 1148–1159. [Google Scholar] [CrossRef]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Apprill, A.; Mcnally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk v2: Memory friendly classification with the genome taxonomy database. Bioinformatics 2022, 38, 5315–5316. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Bunn, A.G. A dendrochronology program library in R (dplR). Dendrochronologia 2008, 26, 115–124. [Google Scholar] [CrossRef]
- Burns, A.R.; Stephens, W.Z.; Stagaman, K.; Wong, S.; Rawls, J.F.; Guillemin, K.; Bohannan, B.J. Contribution of neutral processes to the assembly of gut microbial communities in the zebrafish over host development. ISME J. 2016, 10, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Sloan, W.T.; Woodcock, S.; Lunn, M.; Head, I.M.; Curtis, T.P. Modeling taxa-abundance distributions in microbial communities using environmental sequence data. Microb. Ecol. 2007, 53, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Nearing, J.T.; Douglas, G.M.; Hayes, M.G.; MacDonald, J.; Desai, D.K.; Allward, N.; Jones, C.M.; Wright, R.J.; Dhanani, A.S.; Comeau, A.M.; et al. Microbiome differential abundance methods produce different results across 38 datasets. Nat. Commun. 2022, 13, 342. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Peddada, S.D. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 2020, 11, 3514. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Hashem, A.; Abd Allah, E.F. Bacillus: A biological tool for crop improvement through bio-molecular changes in adverse environments. Front. Physiol. 2017, 8, 667. [Google Scholar] [CrossRef]
- Mujakić, I.; Piwosz, K.; Koblížek, M. Phylum Gemmatimonadota and Its Role in the Environment. Microorganisms 2022, 10, 151. [Google Scholar] [CrossRef]
- Dworkin, M.; Falkow, S.; Rosenberg, E.; Schleifer, K.H.; Stackebrandt, E. The Prokaryotes: A Handbook on the Biology of Bacteria: Archaea. Bacteria: Firmicutes, Actinomycetes; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Grady, E.N.; MacDonald, J.; Liu, L.; Richman, A.; Yuan, Z.C. Current knowledge and perspectives of Paenibacillus: A review. Microb. Cell Factories 2016, 15, 203. [Google Scholar] [CrossRef]
- Ramesh, A.; Sharma, S.K.; Sharma, M.P.; Yadav, N.; Joshi, O.P. Plant Growth-Promoting Traits in Enterobacter cloacae subsp. dissolvens MDSR9 Isolated from Soybean Rhizosphere and its Impact on Growth and Nutrition of Soybean and Wheat Upon Inoculation. Agric. Res. 2014, 3, 53–66. [Google Scholar] [CrossRef]
- Kämpfer, P.; Ruppel, S.; Remus, R. Enterobacter radicincitans sp. nov., a plant growth promoting species of the family Enterobacteriaceae. Syst. Appl. Microbiol. 2005, 28, 213–221. [Google Scholar] [CrossRef]
- Khalifa, A.Y.Z.; Alsyeeh, A.M.; Almalki, M.A.; Saleh, F.A. Characterization of the plant growth promoting bacterium, Enterobacter cloacae MSR1, isolated from roots of non-nodulating Medicago sativa. Saudi J. Biol. Sci. 2016, 23, 79–86. [Google Scholar] [CrossRef]
- Harkes, P.; van Steenbrugge, J.J.M.; van den Elsen, S.J.J.; Suleiman, A.K.A.; de Haan, J.J.; Holterman, M.H.M.; Helder, J. Shifts in the Active Rhizobiome Paralleling Low Meloidogyne chitwoodi Densities in Fields Under Prolonged Organic Soil Management. Front. Plant Sci. 2020, 10, 1697. [Google Scholar] [CrossRef] [PubMed]
- Meier, M.A.; Xu, G.; Lopez-Guerrero, M.G.; Li, G.; Smith, C.; Sigmon, B.; Herr, J.R.; Alfano, J.R.; Ge, Y.; Schnable, J.C.; et al. Association analyses of host genetics, root-colonizing microbes, and plant phenotypes under different nitrogen conditions in maize. eLife 2022, 11, e75790. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.S.; Evans-Fitz Gerald, C.; McElwain, J.C.; Yiotis, C.; Elliott-Kingston, C. How well do you know your growth chambers? Testing for chamber effect using plant traits. Plant Methods 2015, 11, 44. [Google Scholar] [CrossRef] [PubMed]
- Gruber-Dorninger, C.; Pester, M.; Kitzinger, K.; Savio, D.F.; Loy, A.; Rattei, T.; Wagner, M.; Daims, H. Functionally relevant diversity of closely related Nitrospira in activated sludge. ISME J. 2015, 9, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, S.; Bradley, R.L.; Hamel, C.; Beaulieu, C. Do tree-based intercropping systems increase the diversity and stability of soil microbial communities? Agric. Ecosyst. Environ. 2009, 131, 25–31. [Google Scholar] [CrossRef]
- Nottingham, A.T.; Scott, J.J.; Saltonstall, K.; Broders, K.; Montero-Sanchez, M.; Püspök, J.; Bååth, E.; Meir, P. Microbial diversity declines in warmed tropical soil and respiration rise exceed predictions as communities adapt. Nat. Microbiol. 2022, 7, 1650–1660. [Google Scholar] [CrossRef]
- Zegeye, E.K.; Brislawn, C.J.; Farris, Y.; Fansler, S.J.; Hofmockel, K.S.; Jansson, J.K.; Wright, A.T.; Graham, E.B.; Naylor, D.; McClure, R.S.; et al. Selection, Succession, and Stabilization of Soil Microbial Consortia. mSystems 2019, 4, e00055-19. [Google Scholar] [CrossRef]
- Lebeis, S.L.; Paredes, S.H.; Lundberg, D.S.; Breakfield, N.; Gehring, J.; McDonald, M.; Malfatti, S.; del Rio, T.G.; Jones, C.D.; Tringe, S.G.; et al. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science 2015, 349, 860–864. [Google Scholar] [CrossRef]
- Brown, S.P.; Grillo, M.A.; Podowski, J.C.; Heath, K.D. Soil origin and plant genotype structure distinct microbiome compartments in the model legume Medicago truncatula. Microbiome 2020, 8, 139. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Prasad, P.; Das, S.N.; Kalam, S.; Sayyed, R.Z.; Reddy, M.S.; El Enshasy, H. Plant Growth Promoting Rhizobacteria (PGPR) as Green Bioinoculants: Recent Developments, Constraints, and Prospects. Sustainability 2021, 13, 1140. [Google Scholar] [CrossRef]
- Luo, J.; Guo, X.; Tao, Q.; Li, J.; Liu, Y.; Du, Y.; Liu, Y.; Liang, Y.; Li, T. Succession of the composition and co-occurrence networks of rhizosphere microbiota is linked to Cd/Zn hyperaccumulation. Soil Biol. Biochem. 2021, 153, 108120. [Google Scholar] [CrossRef]
- Krishna, M.; Gupta, S.; Delgado-Baquerizo, M.; Morriën, E.; Garkoti, S.C.; Chaturvedi, R.; Ahmad, S. Successional trajectory of bacterial communities in soil are shaped by plant-driven changes during secondary succession. Sci. Rep. 2020, 10, 9864. [Google Scholar] [CrossRef] [PubMed]
- Micallef, S.; Colόn-Carmona, A. Genetic and Developmental Control of Rhizosphere Bacterial Communities. In Molecular Microbial Ecology of the Rhizosphere, 1st ed.; De Bruijn, F.J., Ed.; Wiley: New York, NY, USA, 2013; pp. 257–263. Available online: https://onlinelibrary.wiley.com/doi/10.1002/9781118297674.ch24 (accessed on 19 November 2023).
- Micallef, S.A.; Channer, S.; Shiaris, M.P.; Colón-Carmona, A. Plant age and genotype impact the progression of bacterial community succession in the Arabidopsis rhizosphere. Plant Signal. Behav. 2009, 4, 777–780. [Google Scholar] [CrossRef] [PubMed]
- Monchgesang, S.; Strehmel, N.; Schmidt, S.; Westphal, L.; Taruttis, F.; Muller, E.; Herklotz, S.; Neumann, S.; Scheel, D. Natural variation of root exudates in Arabidopsis thaliana-linking metabolomic and genomic data. Sci. Rep. 2016, 6, 29033. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, C.; Shenton, M.; Kitahara, A.; Wasaki, J.; Oikawa, A.; Cheng, W.; Ikeo, K.; Tawaraya, K. Multiple analysis of root exudates and microbiome in rice (Oryza sativa) under low P conditions. Arch. Microbiol. 2021, 203, 5599–5611. [Google Scholar]
- Huang, X.F.; Chaparro, J.M.; Reardon, K.F.; Zhang, R.; Shen, Q.; Vivanco, J.M. Rhizosphere interactions: Root exudates, microbes, and microbial communities. Botany 2014, 92, 267–275. [Google Scholar] [CrossRef]
- Sun, H.; Jiang, S.; Jiang, C.; Wu, C.; Gao, M.; Wang, Q. A review of root exudates and rhizosphere microbiome for crop production. Env. Sci. Pollut. Res. 2021, 28, 54497–54510. [Google Scholar] [CrossRef]
- Li, Z.; Guo, W.; Mo, C.; Tang, R.; He, L.; Du, L.; Li, M.; Wu, H.; Tang, X.; Huang, Z.; et al. Root Metabolism and Effects of Root Exudates on the Growth of Ralstonia solanacearum and Fusarium moniliforme Were Significantly Different between the Two Genotypes of Peanuts. Genes 2023, 14, 528. [Google Scholar] [CrossRef]
- Badri, D.V.; Vivanco, J.M. Regulation and function of root exudates. Plant Cell Environ. 2009, 32, 666–681. [Google Scholar] [CrossRef]
- Cotton, T.E.A.; Pétriacq, P.; Cameron, D.D.; Meselmani, M.A.; Schwarzenbacher, R.; Rolfe, S.A.; Ton, J. Metabolic regulation of the maize rhizobiome by benzoxazinoids. ISME J. 2019, 13, 1647–1658. [Google Scholar] [CrossRef]
- Cadot, S.; Guan, H.; Bigalke, M.; Walser, J.C.; Jander, G.; Erb, M.; van der Heijden, M.G.; Schlaeppi, K. Specific and conserved patterns of microbiota-structuring by maize benzoxazinoids in the field. Microbiome 2021, 9, 103. [Google Scholar] [CrossRef]
- Korenblum, E.; Dong, Y.; Szymanski, J.; Panda, S.; Jozwiak, A.; Massalha, H.; Meir, S.; Rogachev, I.; Aharoni, A. Rhizosphere microbiome mediates systemic root metabolite exudation by root-to-root signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 3874–3883. [Google Scholar] [CrossRef] [PubMed]
- Rudrappa, T.; Czymmek, K.J.; Paré, P.W.; Bais, H.P. Root-Secreted Malic Acid Recruits Beneficial Soil Bacteria. Plant Physiol. 2008, 148, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant–microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef]
- Gu, S.; Wei, Z.; Shao, Z.; Friman, V.P.; Cao, K.; Yang, T.; Kramer, J.; Wang, X.; Li, M.; Mei, X.; et al. Competition for iron drives phytopathogen control by natural rhizosphere microbiomes. Nat. Microbiol. 2020, 5, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.C.; Chang, Y.Y.; Hussain, M.; Lu, B.; Zhang, J.P.; Song, X.B.; Lei, X.S.; Pei, D. Soil Chemical and Microbiological Properties Are Changed by Long-Term Chemical Fertilizers That Limit Ecosystem Functioning. Microorganisms 2020, 8, 694. [Google Scholar] [CrossRef]
- Xu, Q.; Ling, N.; Chen, H.; Duan, Y.; Wang, S.; Shen, Q.; Vandenkoornhuyse, P. Long-Term Chemical-Only Fertilization Induces a Diversity Decline and Deep Selection on the Soil Bacteria. mSystems 2020, 5, e00337-20. [Google Scholar] [CrossRef]
- Cardinale, M.; Ratering, S.; Suarez, C.; Zapata Montoya, A.M.; Geissler-Plaum, R.; Schnell, S. Paradox of plant growth promotion potential of rhizobacteria and their actual promotion effect on growth of barley (Hordeum vulgare L.) under salt stress. Microbiol. Res. 2015, 181, 22–32. [Google Scholar] [CrossRef]
- Meena, M.; Swapnil, P.; Divyanshu, K.; Kumar, S.; Harish; Tripathi, Y.N.; Zehra, A.; Marwal, A.; Upadhyay, R.S. PGPR-mediated induction of systemic resistance and physiochemical alterations in plants against the pathogens: Current perspectives. J. Basic Microbiol. 2020, 60, 828–861. [Google Scholar] [CrossRef] [PubMed]
- Kamilova, F.; Okon, Y.; de Weert, S.; Hora, K. Commercialization of Microbes: Manufacturing, Inoculation, Best Practice for Objective Field Testing, and Registration. In Principles of Plant-Microbe Interactions: Microbes for Sustainable Agriculture; Lugtenberg, B., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 319–327. [Google Scholar] [CrossRef]
- Dubey, S.; Sharma, S. Rhizospheric Engineering by Plant-Mediated Indirect Selection of Microbiome for Agricultural Sustainability. Crit. Rev. Plant Sci. 2021, 40, 379–397. [Google Scholar] [CrossRef]
- Wright, R.J.; Gibson, M.I.; Christie-Oleza, J.A. Understanding microbial community dynamics to improve optimal microbiome selection. Microbiome 2019, 7, 85. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shankar, N.; Shetty, P.; Melo, T.C.; Kesseli, R. Multi-Generation Ecosystem Selection of Rhizosphere Microbial Communities Associated with Plant Genotype and Biomass in Arabidopsis thaliana. Microorganisms 2023, 11, 2932. https://doi.org/10.3390/microorganisms11122932
Shankar N, Shetty P, Melo TC, Kesseli R. Multi-Generation Ecosystem Selection of Rhizosphere Microbial Communities Associated with Plant Genotype and Biomass in Arabidopsis thaliana. Microorganisms. 2023; 11(12):2932. https://doi.org/10.3390/microorganisms11122932
Chicago/Turabian StyleShankar, Nachiket, Prateek Shetty, Tatiana C. Melo, and Rick Kesseli. 2023. "Multi-Generation Ecosystem Selection of Rhizosphere Microbial Communities Associated with Plant Genotype and Biomass in Arabidopsis thaliana" Microorganisms 11, no. 12: 2932. https://doi.org/10.3390/microorganisms11122932
APA StyleShankar, N., Shetty, P., Melo, T. C., & Kesseli, R. (2023). Multi-Generation Ecosystem Selection of Rhizosphere Microbial Communities Associated with Plant Genotype and Biomass in Arabidopsis thaliana. Microorganisms, 11(12), 2932. https://doi.org/10.3390/microorganisms11122932