
Associations of Microbial Diversity with Age and Other Clinical Variables among Pediatric Chronic Rhinosinusitis (CRS) Patients
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Enrollment and Sample Collection
2.2. Data Analysis
3. Results
3.1. Participant Characteristics
3.2. Sinus Culture Results
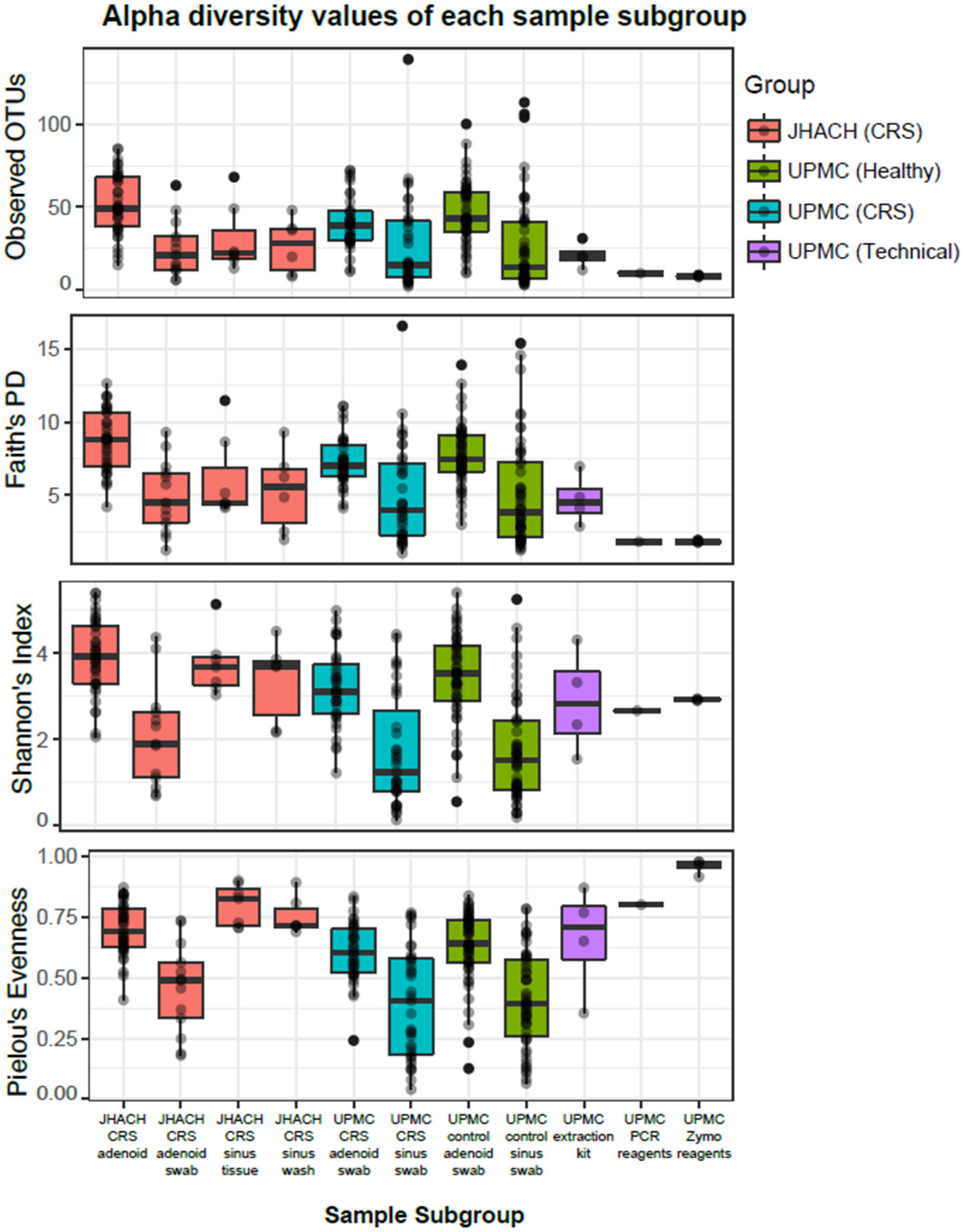
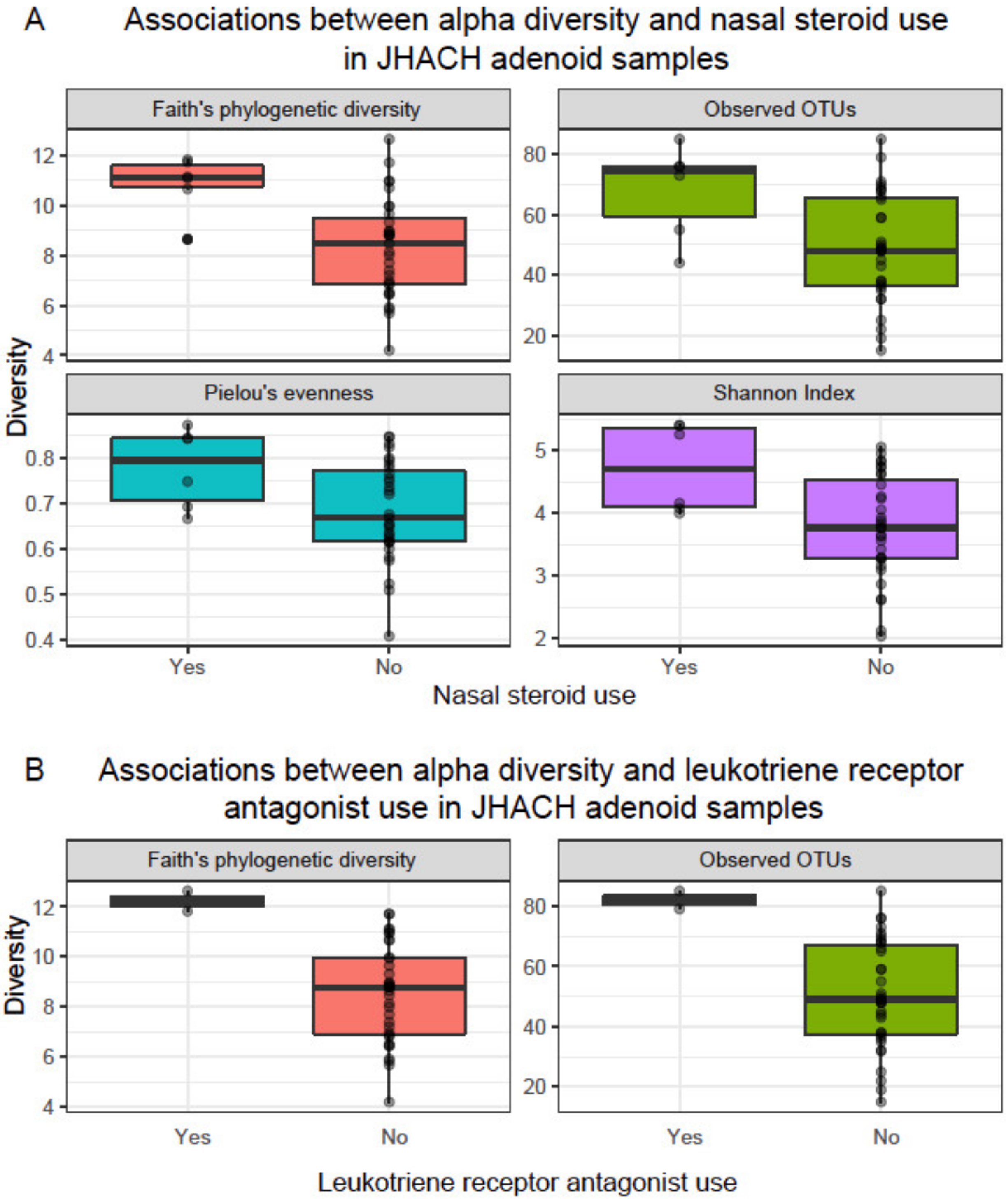
3.3. Alpha Diversity
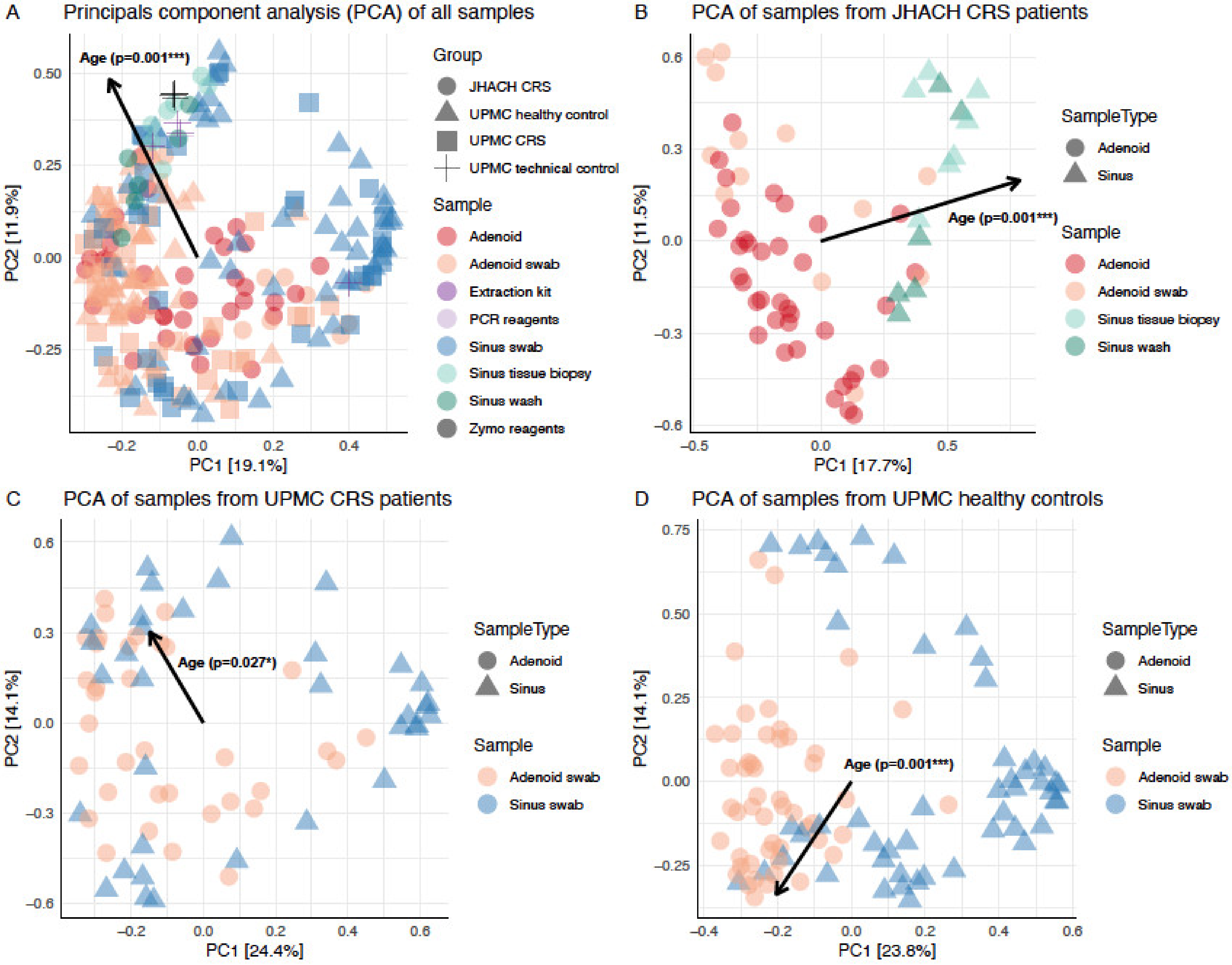
3.4. Beta Diversity
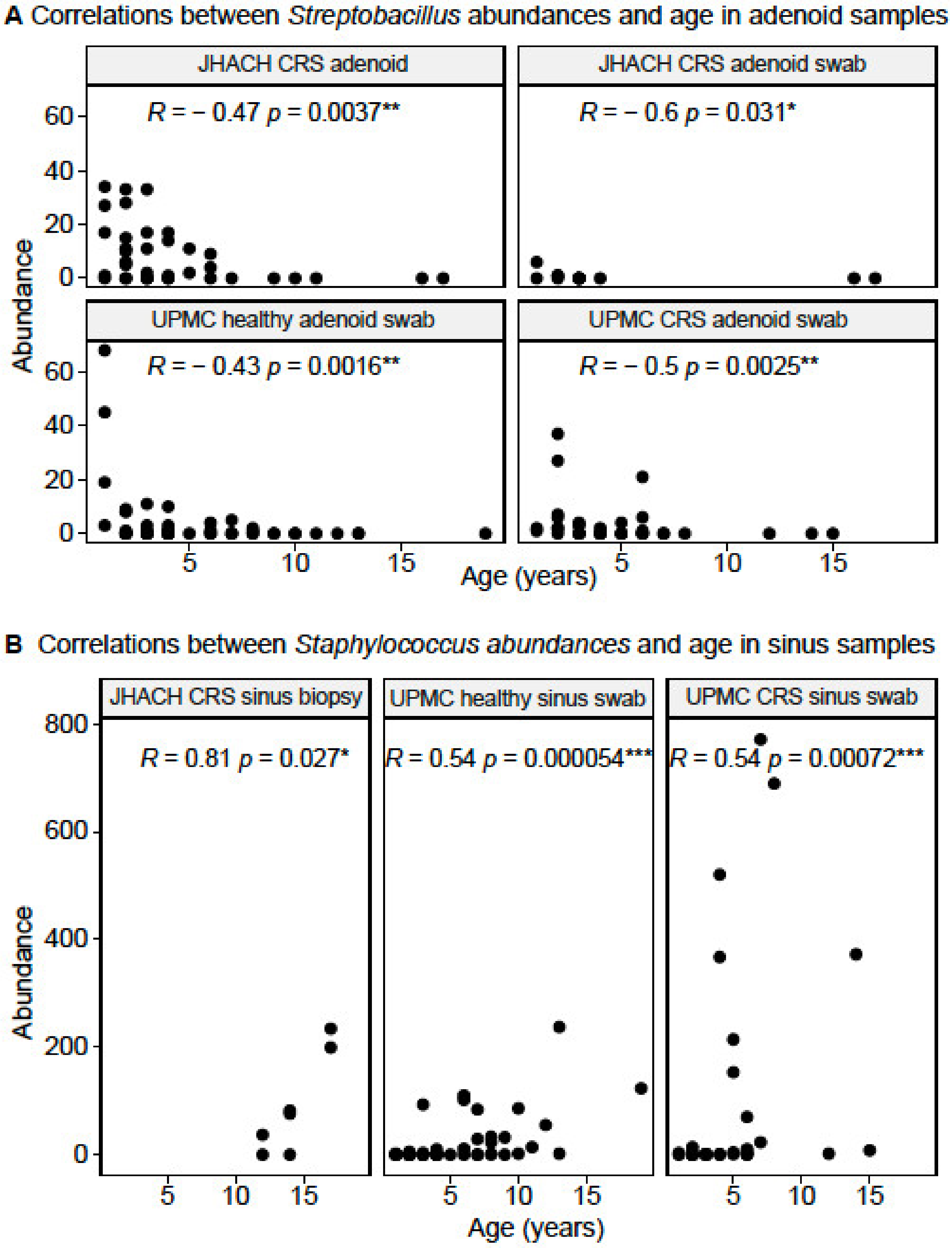
3.5. Taxonomic Composition in Adenoid-Derived Samples
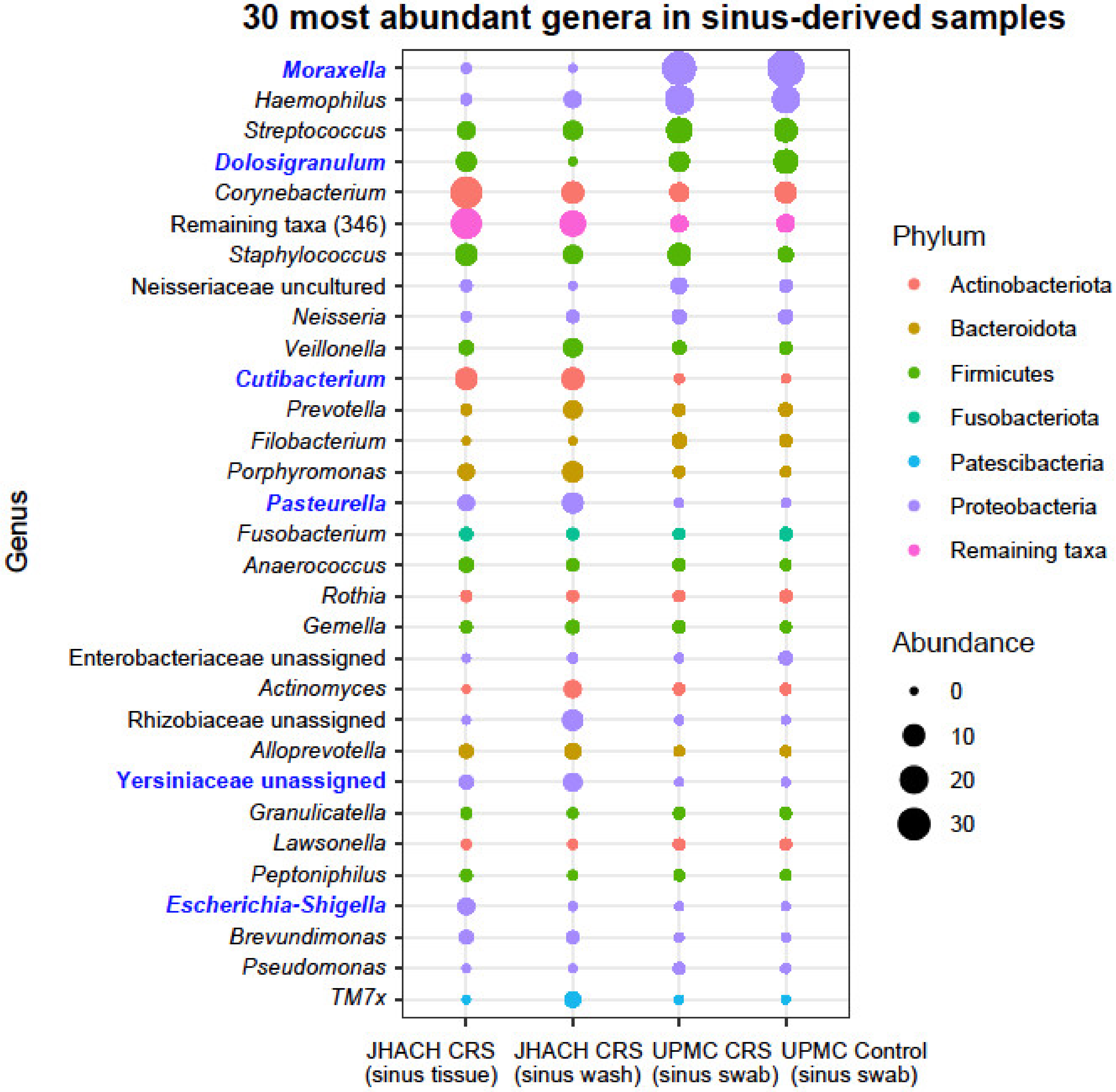
3.6. Taxonomic Composition in Sinus-Derived Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hopp, R.J. Do Adult Forms of Chronic Rhinosinusitis Exist in Children and Adolescents? Sinusitis 2017, 2, 7. [Google Scholar] [CrossRef]
- Hopp, R.J. Pediatric chronic rhinosinusitis: Unmet needs. Sinusitis 2020, 4, 2–7. [Google Scholar] [CrossRef]
- Snidvongs, K.; Sangubol, M.; Poachanukoon, O. Pediatric versus adult chronic rhinosinusitis. Curr. Allergy Asthma Rep. 2020, 20, 29. [Google Scholar] [CrossRef] [PubMed]
- Hamilos, D.L. Pediatric chronic rhinosinusitis. Am. J. Rhinol. Allergy 2015, 29, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat, A.R. Chronic rhinosinusitis. Am. Fam. Physician 2017, 96, 500–506. [Google Scholar]
- Chandy, Z.; Ference, E.; Lee, J.T. Clinical guidelines on chronic rhinosinusitis in children. Curr. Allergy Asthma Rep. 2019, 19, 14. [Google Scholar] [CrossRef]
- Slack, R.; Bates, G. Functional endoscopic sinus surgery. Am. Fam. Physician 1998, 58, 707–718. [Google Scholar]
- Foreman, A.; Boase, S.; Psaltis, A.; Wormald, P.J. Role of bacterial and fungal biofilms in chronic rhinosinusitis. Curr. Allergy Asthma Rep. 2012, 12, 127–135. [Google Scholar] [CrossRef]
- Hoggard, M.; Wagner Mackenzie, B.; Jain, R.; Taylor, M.W.; Biswas, K.; Douglas, R.G. Chronic rhinosinusitis and the evolving understanding of microbial ecology in chronic inflammatory mucosal disease. Clin. Microbiol. Rev. 2017, 30, 321–348. [Google Scholar] [CrossRef]
- Drago, L.; Pignataro, L.; Torretta, S. Microbiological aspects of acute and chronic pediatric rhinosinusitis. J. Clin. Med. 2019, 8, 149. [Google Scholar] [CrossRef]
- Hsin, C.H.; Su, M.C.; Tsao, C.H.; Chuang, C.Y.; Liu, C.M. Bacteriology and antimicrobial susceptibility of pediatric chronic rhinosinusitis: A 6-year result of maxillary sinus punctures. Am. J. Otolaryngol. 2010, 31, 145–149. [Google Scholar] [CrossRef]
- Coticchia, J.; Zuliani, G.; Coleman, C.; Carron, M.; Gurrola, J., II; Haupert, M.; Berk, R. Biofilm surface area in the pediatric nasopharynx: Chronic rhinosinusitis vs. obstructive sleep apnea. Arch. Otolaryngol.-Head Neck Surg. 2007, 133, 110–114. [Google Scholar] [CrossRef]
- Bugari, R.A.; Baschir, A.S.; Turcin, L.A.; Chioreanu, A.; Mihali, C.V.; Ilie, A.C.; Jompan, A.; Balasoiu, M. Adenoidal bacterial biofilm in pediatric rhinosinusitis. Rom. J. Morphol. Embryol. 2021, 62, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Kania, R.E.; Lamers, G.E.; Vonk, M.J.; Dorpmans, E.; Struik, J.; Tran Ba Huy, P.; Hiemstra, P.; Bloemberg, G.V.; Grote, J.J. Characterization of mucosal biofilms on human adenoid tissues. Laryngoscope 2008, 118, 128–134. [Google Scholar] [CrossRef]
- Pasha, M.A. State-of-the-art adult chronic rhinosinusitis microbiome: Perspective for future studies in pediatrics. Sinusitis 2018, 3, 1. [Google Scholar] [CrossRef]
- Hauser, L.J.; Feazel, L.M.; Ir, D.; Fang, R.; Wagner, B.D.; Robertson, C.E.; Frank, D.N.; Ramakrishnan, V.R. Sinus culture poorly predicts resident microbiota. Int. Forum Allergy Rhinol. 2015, 5, 3–9. [Google Scholar] [CrossRef]
- Joss, T.V.; Burke, C.M.; Hudson, B.J.; Darling, A.E.; Forer, M.; Alber, D.G.; Charles, I.G.; Stow, N.W. Bacterial communities vary between sinuses in chronic rhinosinusitis patients. Front. Microbiol. 2015, 6, 1532. [Google Scholar] [CrossRef]
- Koeller, K.; Herlemann, D.P.R.; Schuldt, T.; Ovari, A.; Guder, E.; Podbielski, A.; Kreikemeyer, B.; Olzowy, B. Microbiome and culture based analysis of chronic rhinosinusitis compared to healthy sinus mucosa. Front. Microbiol. 2018, 9, 643. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.C.; Yeh, Y.T.; Lin, C.C.; Huang, C.H.; Liu, H.C.; Chiang, C.P. Clinical detection of chronic rhinosinusitis through next-generation sequencing of the oral microbiota. Microorganisms 2020, 8, 959. [Google Scholar] [CrossRef] [PubMed]
- Abreu, N.A.; Nagalingam, N.A.; Song, Y.; Roediger, F.C.; Pletcher, S.D.; Goldberg, A.N.; Lynch, S.V. Sinus microbiome diversity depletion and Corynebacterium tuberculostearicum enrichment mediates rhinosinusitis. Sci. Transl. Med. 2012, 4, 151ra124. [Google Scholar] [CrossRef] [PubMed]
- Hoggard, M.; Biswas, K.; Zoing, M.; Wagner Mackenzie, B.; Taylor, M.W.; Douglas, R.G. Evidence of microbiota dysbiosis in chronic rhinosinusitis. Int. Forum Allergy Rhinol. 2017, 7, 230–239. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, I.; Wittouck, S.; Martens, K.; Claes, J.; Jorissen, M.; Steelant, B.; van den Broek, M.F.L.; Seys, S.F.; Hellings, P.W.; Vanderveken, O.M.; et al. Anterior nares diversity and pathobionts represent sinus microbiome in chronic rhinosinusitis. mSphere 2019, 4, e00532-19. [Google Scholar] [CrossRef]
- Stapleton, A.L.; Shaffer, A.D.; Morris, A.; Li, K.; Fitch, A.; Methe, B.A. The microbiome of pediatric patients with chronic rhinosinusitis. Int. Forum Allergy Rhinol. 2021, 11, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Knight, R.; Vrbanac, A.; Taylor, B.C.; Aksenov, A.; Callewaert, C.; Debelius, J.; Gonzalez, A.; Kosciolek, T.; McCall, L.I.; McDonald, D.; et al. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 2018, 16, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Janssen, S.; McDonald, D.; Gonzalez, A.; Navas-Molina, J.A.; Jiang, L.; Xu, Z.Z.; Winker, K.; Kado, D.M.; Orwoll, E.; Manary, M.; et al. Phylogenetic placement of exact amplicon sequences improves associations with clinical information. mSystems 2018, 3, e00021-18. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- The R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Andersen, K.S.; Kirkegaard, R.H.; Karst, S.M.; Albertsen, M. ampvis2: An R package to analyse and visualise 16S rRNA amplicon data. bioRxiv 2018, 299537. [Google Scholar] [CrossRef]
- Legendre, P.; Gallagher, E.D. Ecologically meaningful transformations for ordination of species data. Oecologia 2001, 129, 271–280. [Google Scholar] [CrossRef]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbo, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef] [PubMed]
- Mallick, H.; Rahnavard, A.; McIver, L.J.; Ma, S.; Zhang, Y.; Nguyen, L.H.; Tickle, T.L.; Weingart, G.; Ren, B.; Schwager, E.H.; et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 2021, 17, e1009442. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat.Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Davis, M.L.; Austin, C.; Messmer, B.L.; Nichols, W.K.; Bonin, A.; Bennett, M.J. IFCC-standardized pediatric reference intervals for 10 serum proteins using the Beckman Array 360 system©. Clin. Biochem. 1996, 29, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Willis, A.D. Rarefaction, alpha diversity, and statistics. Front. Microbiol. 2019, 10, 2407. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, R.R.; Kingdom, T.T.; Hwang, P.H.; Smith, T.L.; Alt, J.A.; Baroody, F.M.; Batra, P.S.; Bernal-Sprekelsen, M.; Bhattacharyya, N.; Chandra, R.K.; et al. International consensus statement on allergy and rhinology: Rhinosinusitis. Int. Forum Allergy Rhinol. 2016, 6 (Suppl. S1), S22–S209. [Google Scholar] [CrossRef]
- Mancuso, R.F. Pediatric allergic fungal sinusitis: A clinical review. Pediatr. Ann. 2021, 50, e297–e303. [Google Scholar] [CrossRef]
- Ramakrishnan, V.R.; Holt, J.; Nelson, L.F.; Ir, D.; Robertson, C.E.; Frank, D.N. Determinants of the nasal microbiome: Pilot study of effects of intranasal medication use. Allergy Rhinol. 2018, 9, 2152656718789519. [Google Scholar] [CrossRef]
- Melvin, T.-A.N.; Patel, A.A. Pharmacotherapy for allergic rhinitis. Otolaryngol. Clin. N. Am. 2011, 44, 727–739. [Google Scholar] [CrossRef]
- Wastyk, H.C.; Fragiadakis, G.K.; Perelman, D.; Dahan, D.; Merrill, B.D.; Yu, F.B.; Topf, M.; Gonzalez, C.G.; Van Treuren, W.; Han, S.; et al. Gut-microbiota-targeted diets modulate human immune status. Cell 2021, 184, 4137–4153.e14. [Google Scholar] [CrossRef]
- Hussien, H.A.; Habieb, M.S.; Hamdan, A.M. Evaluation of serum total immunoglobulin E, interleukin-17 and pentraxin-3 as biomarkers for chronic rhinosinusitis with nasal polyposis. Am. J. Rhinol. Allergy 2021, 35, 640–646. [Google Scholar] [CrossRef]
- Kim, S.K.; Hong, S.J.; Pak, K.H.; Hong, S.M. Analysis of the microbiome in the adenoids of Korean Children with otitis media with effusion. J. Int. Adv. Otol. 2019, 15, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Gao, W.; Yu, C.; Li, J.; Yu, F.; Xia, M.; Liang, J.; Shi, J.; Lai, Y. Age-associated changes of nasal bacterial microbiome in patients with chronic rhinosinusitis. Front. Cell Infect. Microbiol. 2022, 12, 786481. [Google Scholar] [CrossRef] [PubMed]
- Westphal, C.; Gorlich, D.; Kampmeier, S.; Herzog, S.; Braun, N.; Hitschke, C.; Mellmann, A.; Peters, G.; Kahl, B.C.; Staphylococcal, C.F.S.G. Antibiotic treatment and age are associated with Staphylococcus aureus carriage profiles during oersistence in the airways of cystic fibrosis patients. Front. Microbiol. 2020, 11, 230. [Google Scholar] [CrossRef] [PubMed]
- Hurst, J.H.; McCumber, A.W.; Aquino, J.N.; Rodriguez, J.; Heston, S.M.; Lugo, D.J.; Rotta, A.T.; Turner, N.A.; Pfeiffer, T.S.; Gurley, T.C.; et al. Age-related changes in the nasopharyngeal microbiome are associated with SARS-CoV-2 infection and symptoms among children, adolescents, and young adults. Clin. Infect. Dis. 2022, 75, e928–e937. [Google Scholar] [CrossRef] [PubMed]
- Drevinek, P.; Mahenthiralingam, E. Burkholderia cenocepacia in cystic fibrosis: Epidemiology and molecular mechanisms of virulence. Clin. Microbiol. Infect. 2010, 16, 821–830. [Google Scholar] [CrossRef]
- Riskumaki, M.; Tessas, I.; Ottman, N.; Suomalainen, A.; Werner, P.; Karisola, P.; Lauerma, A.; Ruokolainen, L.; Karkman, A.; Wisgrill, L.; et al. Interplay between skin microbiota and immunity in atopic individuals. Allergy 2021, 76, 1280–1284. [Google Scholar] [CrossRef] [PubMed]
- Le Guern, A.S.; Savin, C.; Angermeier, H.; Bremont, S.; Clermont, D.; Muhle, E.; Orozova, P.; Najdenski, H.; Pizarro-Cerda, J. Yersinia artesiana sp. nov., Yersinia proxima sp. nov., Yersinia alsatica sp. nov., Yersina vastinensis sp. nov., Yersinia thracica sp. nov. and Yersinia occitanica sp. nov., isolated from humans and animals. Int. J. Syst. Evol. Microbiol. 2020, 70, 5363–5372. [Google Scholar] [CrossRef]
- La Scola, B.; Fournier, P.E.; Raoult, D. Burden of emerging anaerobes in the MALDI-TOF and 16S rRNA gene sequencing era. Anaerobe 2011, 17, 106–112. [Google Scholar] [CrossRef]
- Corvec, S. Clinical and biological features of Cutibacterium (formerly Propionibacterium) avidum, an underrecognized microorganism. Clin. Microbiol. Rev. 2018, 31, e00064-17. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Adenoidectomy Patients n = 40 | FESS Patients n = 5 |
|---|---|---|
| Sex | ||
| Female, n (%) | 14 (35) | 3 (60) |
| Male, n (%) | 26 (65) | 2 (40) |
| Age, mean ± SD (years) | 4.3 ± 3.7 | 13 ± 3.3 |
| Sinus culture | ||
| Yes, n (%) | 15 (37.5) | 5 (100) |
| No, n (%) | 25 (62.5) | 0 (0) |
| Adenoid tissue sequenced | ||
| Yes, n (%) | 40 (100) | - |
| Adenoid swab sequenced | ||
| Yes, n (%) | 14 (35) | - |
| No, n (%) | 26 (65) | - |
| Sinus biopsy sequenced | ||
| Yes, n (%) | - | 5 (100) |
| Sinus wash sequenced | ||
| Yes, n (%) | - | 5 (100) |
| Atopy | ||
| Asthma | ||
| Yes, n (%) | 1 (2.5) | 0 (0) |
| No, n (%) | 39 (97.5) | 5 (100) |
| Allergic rhinitis | ||
| Yes, n (%) | 14 (35) | 2 (40) |
| No, n (%) | 26 (65) | 3 (60) |
| Serum IgE test | ||
| Yes, n (%) | 7 (17.5) | 5 (100) |
| No, n (%) | 33 (82.5) | 0 (0) |
| Mean ± SD (IU/mL) | 258.8 ± 496.3 | 605.2 ± 903.8 |
| Serum IgG test | ||
| Yes, n (%) | 2 (5) | 4 (80) |
| No, n (%) | 38 (95) | 1 (20) |
| Mean ± SD (mg/dL) | 647.5 ± 304.8 | 1178.3 ± 208.7 |
| Serum IgA test | ||
| Yes, n (%) | 2 (5) | 4 (80) |
| No, n (%) | 38 (95) | 1 (20) |
| Mean ± SD (mg/dL) | 60.5 ± 60.1 | 148 ± 66 |
| Serum IgM test | ||
| Yes, n (%) | 2 (5) | 4 (80) |
| No, n (%) | 38 (95) | 1 (20) |
| Mean ± SD (mg/dL) | 75.5 ± 10.6 | 173 ± 55 |
| Medications | ||
| Antibiotic | ||
| Yes, n (%) | 13 (32.5) | 3 (60) |
| No, n (%) | 27 (67.5) | 2 (40) |
| Antihistamine | ||
| Yes, n (%) | 15 (37.5) | 1 (20) |
| No, n (%) | 25 (62.5) | 4 (80) |
| Inhaled steroid | ||
| Yes, n (%) | 4 (10) | 0 (0) |
| No, n (%) | 36 (90) | 5 (100) |
| Inhaled β2 antagonist | ||
| Yes, n (%) | 4 (10) | 0 (0) |
| No, n (%) | 36 (90) | 5 (100) |
| Nasal steroid | ||
| Yes, n (%) | 6 (15) | 1 (20) |
| No, n (%) | 34 (85) | 4 (80) |
| Leukotriene receptor antagonist | ||
| Yes, n (%) | 2 (5) | 0 (0) |
| No, n (%) | 38 (95) | 5 (100) |
| Proton pump inhibitor | ||
| Yes, n (%) | 0 (0) | 0 (0) |
| No, n (%) | 40 (100) | 5 (100) |
| Species, n (%) | Adenoidectomy Patients n = 15 | FESS Patients n = 5 |
|---|---|---|
| Streptococcus pneumoniae | 6 (40.0) | 1 (20) |
| Moraxella catarrhalis | 6 (40.0) | 0 (0.0) |
| Haemophilus influenza | 5 (33.3) | 0 (0.0) |
| Staphylococcus aureus | 2 (13.3) | 0 (0.0) |
| Streptococcus pyogenes | 1 (6.7) | 0 (0.0) |
| Pseudomonas aeruginosa | 1 (6.7) | 0 (0.0) |
| Gram-positive cocci | 1 (6.7) | 0 (0.0) |
| Curvularia species | 0 (0.0) | 1 (20) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, S.J.; Jithpratuck, W.; Wasylik, K.; Sriaroon, P.; Dishaw, L.J. Associations of Microbial Diversity with Age and Other Clinical Variables among Pediatric Chronic Rhinosinusitis (CRS) Patients. Microorganisms 2023, 11, 422. https://doi.org/10.3390/microorganisms11020422
Lim SJ, Jithpratuck W, Wasylik K, Sriaroon P, Dishaw LJ. Associations of Microbial Diversity with Age and Other Clinical Variables among Pediatric Chronic Rhinosinusitis (CRS) Patients. Microorganisms. 2023; 11(2):422. https://doi.org/10.3390/microorganisms11020422
Chicago/Turabian StyleLim, Shen Jean, Warit Jithpratuck, Kathleen Wasylik, Panida Sriaroon, and Larry J. Dishaw. 2023. "Associations of Microbial Diversity with Age and Other Clinical Variables among Pediatric Chronic Rhinosinusitis (CRS) Patients" Microorganisms 11, no. 2: 422. https://doi.org/10.3390/microorganisms11020422