
The microRNA Cargo of Human Vaginal Extracellular Vesicles Differentiates Parasitic and Pathobiont Infections from Colonization by Homeostatic Bacteria

Abstract
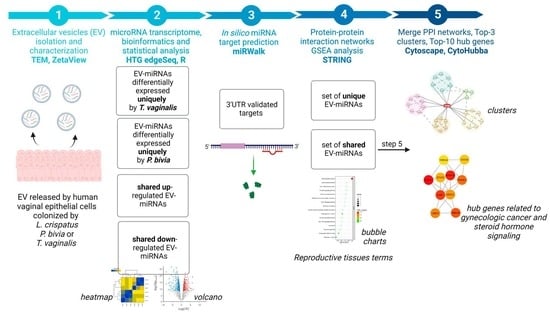
1. Introduction
2. Materials and Methods
2.1. Experimental Model
2.2. Small Extracellular Vesicles Isolation and Characterization
2.3. Whole Human miRNA Transcriptome Profiling
2.4. miRNA Transcriptome Bioinformatics and Statistical Analysis
2.5. miRNA-Target Genes Prediction and Gene Set Enrichment Analysis
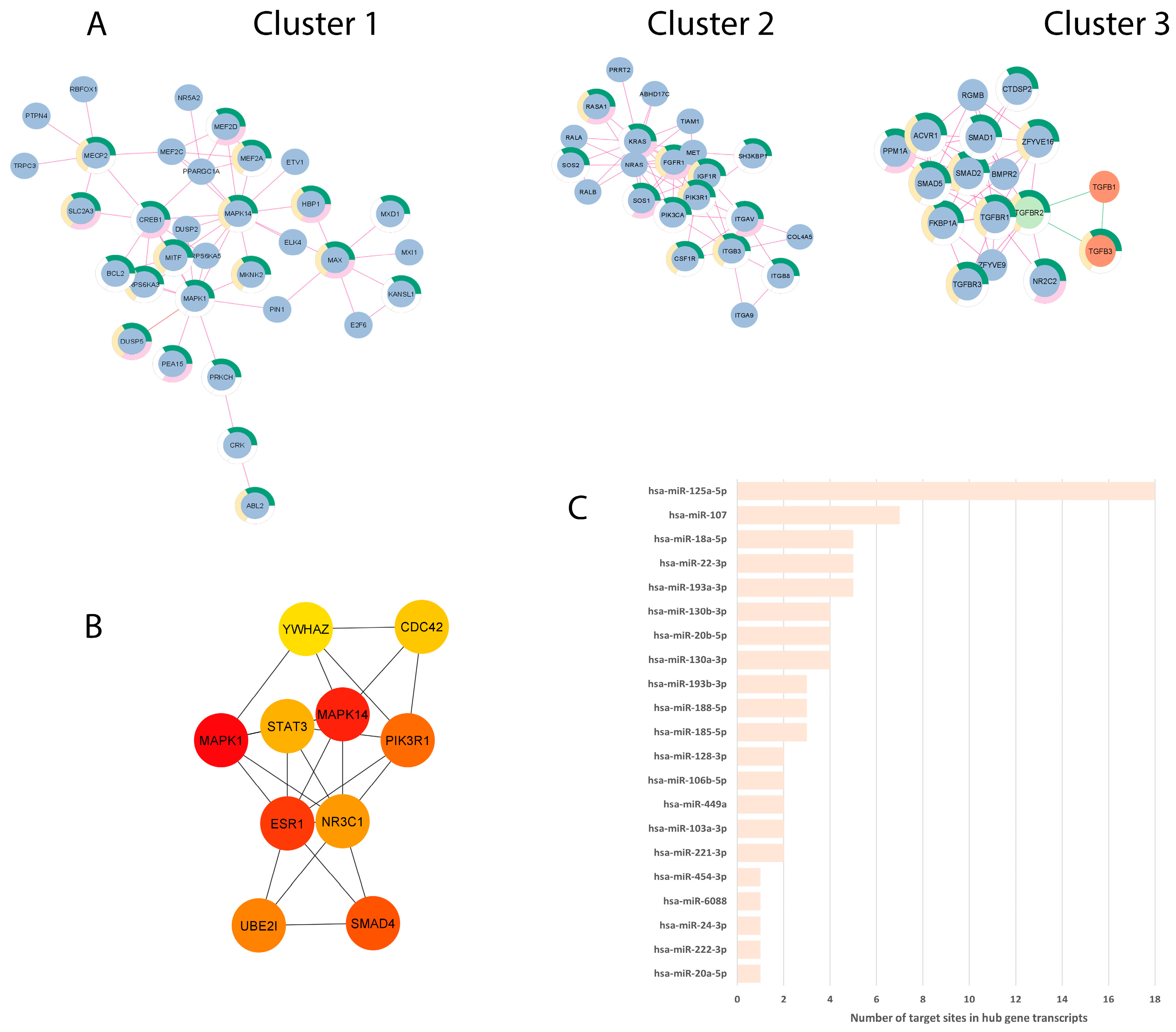
2.6. Protein-Protein Interaction Network Analysis
3. Results
3.1. Non-Colonized and Colonized Human Vaginal Epithelial Cells Release Exosomes
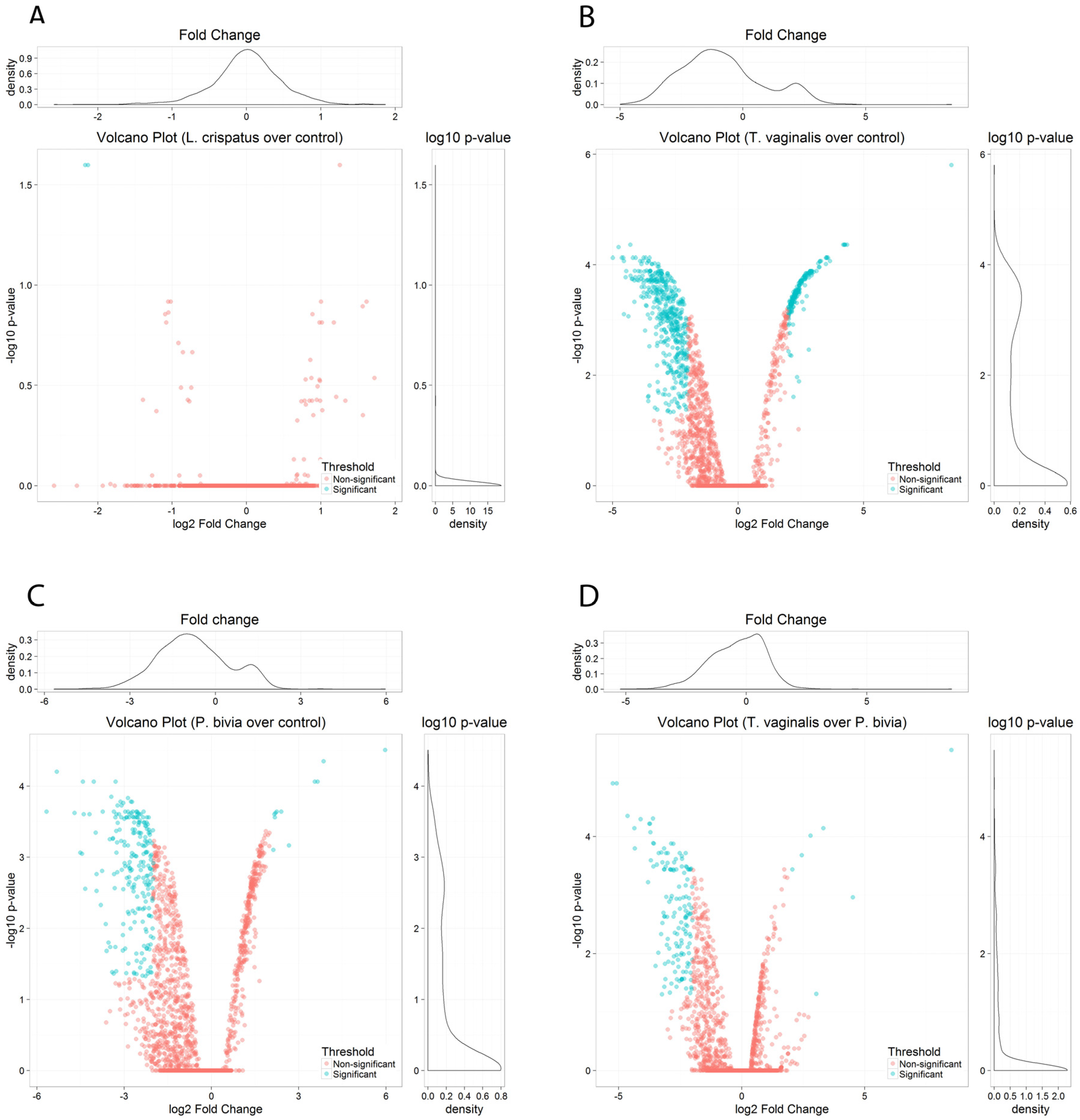
3.2. miRNAs-Containing Extracellular Vesicles from Colonized Human Vaginal Epithelial Cells Identified Pathogenic and Healthy Signatures
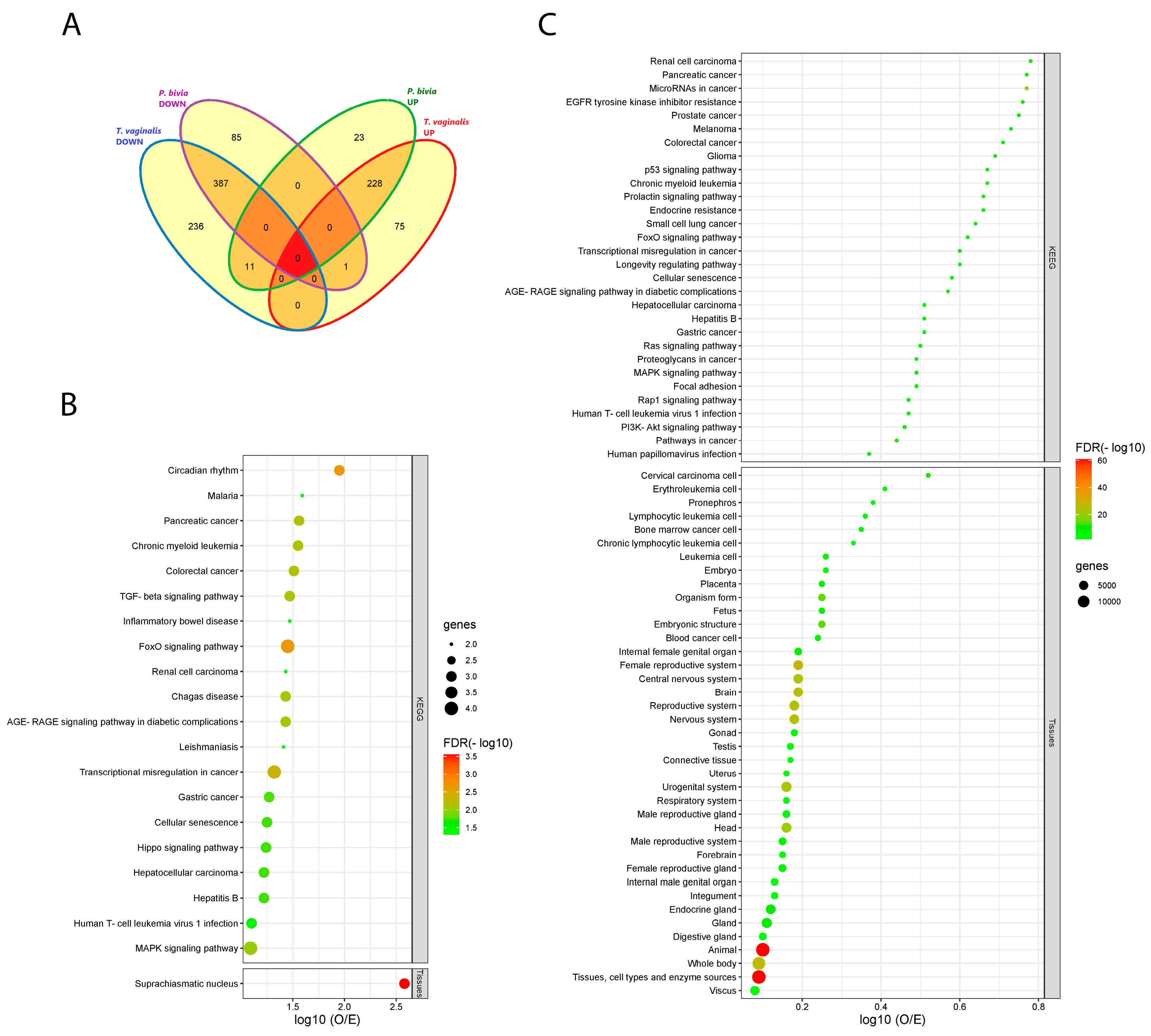
3.3. Vaginal Epithelial Cell Colonization by T. vaginalis and the BV-Pathobiont Identified EV-miRNA Targeted Genes and Pathways Associated with Cancer, Viral Infections, and Potential Reproductive Tract Tissue Recipients
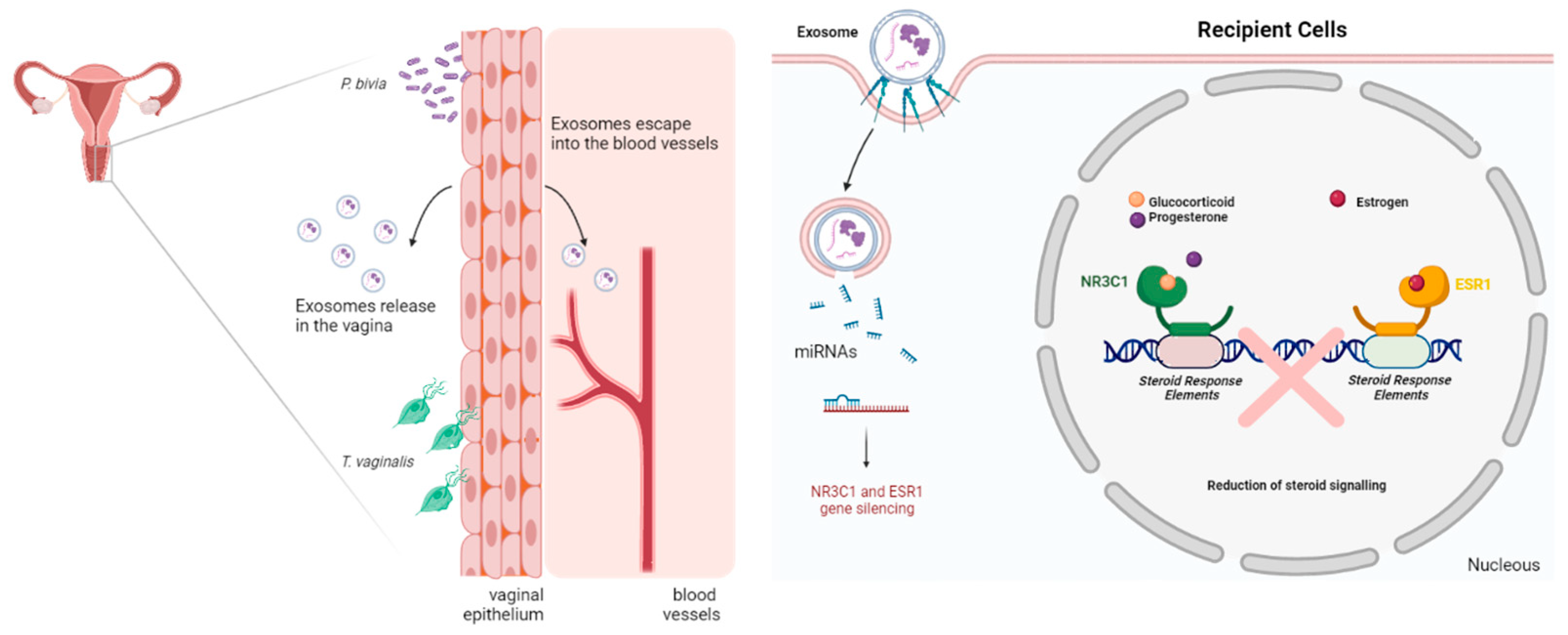
3.4. miRNA Dysregulation by Parasitic and BV-Associated Organisms Targets Steroid Hormone Receptor Signaling and Pathways Associated with Cancer, Viral Infections, and Potential Reproductive Tract Tissue Recipients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shahab, M.; Shahab, N. Coevolution of the Human Host and Gut Microbiome: Metagenomics of Microbiota. Cureus 2022, 14, e26310. [Google Scholar] [CrossRef]
- Kapsetaki, S.E.; Marquez Alcaraz, G.; Maley, C.C.; Whisner, C.M.; Aktipis, A. Diet, Microbes, and Cancer Across the Tree of Life: A Systematic Review. Curr. Nutr. Rep. 2022, 11, 508–525. [Google Scholar] [CrossRef] [PubMed]
- González Cordero, E.M.; Cuevas-Budhart, M.A.; Pérez Morán, D.; Trejo Villeda, M.A.; Gomez-Del-Pulgar G-Madrid, M. Relationship Between the Gut Microbiota and Alzheimer’s Disease: A Systematic Review. J. Alzheimers Dis. 2022, 87, 519–528. [Google Scholar] [CrossRef]
- Consales, A.; Cerasani, J.; Sorrentino, G.; Morniroli, D.; Colombo, L.; Mosca, F.; Giannì, M.L. The hidden universe of human milk microbiome: Origin, composition, determinants, role, and future perspectives. Eur. J. Pediatr. 2022, 181, 1811–1820. [Google Scholar] [CrossRef]
- Chen, C.; Song, X.; Wei, W.; Zhong, H.; Dai, J.; Lan, Z.; Li, F.; Yu, X.; Feng, Q.; Wang, Z.; et al. The microbiota continuum along the female reproductive tract and its relation to uterine-related diseases. Nat. Commun. 2017, 8, 875. [Google Scholar] [CrossRef]
- Onderdonk, A.B.; Delaney, M.L.; Fichorova, R.N. The Human Microbiome during Bacterial Vaginosis. Clin. Microbiol. Rev. 2016, 29, 223–238. [Google Scholar] [CrossRef]
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.K.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O.; et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 2011, 108, 4680–4687. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The Human Microbiome Project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Plesniarski, A.; Siddik, A.B.; Su, R.C. The Microbiome as a Key Regulator of Female Genital Tract Barrier Function. Front. Cell Infect. Microbiol. 2021, 11, 790627. [Google Scholar] [CrossRef]
- Witkin, S.; Linhares, I. Why do lactobacilli dominate the human vaginal microbiota? BJOG Int. J. Obstet. Gynaecol. 2017, 124, 606–611. [Google Scholar] [CrossRef]
- Zozaya-Hinchliffe, M.; Lillis, R.; Martin, D.H.; Ferris, M.J. Quantitative PCR Assessments of Bacterial Species in Women with and without Bacterial Vaginosis. J. Clin. Microbiol. 2010, 48, 1812–1819. [Google Scholar] [CrossRef]
- Delaney, M.L.; Onderdonk, A.B. Nugent score related to vaginal culture in pregnant women. Obstet. Gynecol. 2001, 98, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Anahtar, M.N.; Byrne, E.H.; Doherty, K.E.; Bowman, B.A.; Yamamoto, H.S.; Soumillon, M.; Padavattan, N.; Ismail, N.; Moodley, A.; Sabatini, M.E.; et al. Cervicovaginal bacteria are a major modulator of host inflammatory responses in the female genital tract. Immunity 2015, 42, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Brusselaers, N.; Shrestha, S.; van de Wijgert, J.; Verstraelen, H. Vaginal dysbiosis and the risk of human papillomavirus and cervical cancer: Systematic review and meta-analysis. Am. J. Obs. Gynecol. 2019, 221, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Alimena, S.; Davis, J.; Fichorova, R.N.; Feldman, S. The vaginal microbiome: A complex milieu affecting risk of human papillomavirus persistence and cervical cancer. Curr. Probl. Cancer 2022, 46, 100877. [Google Scholar] [CrossRef] [PubMed]
- Atashili, J.; Poole, C.; Ndumbe, P.M.; Adimora, A.A.; Smith, J.S. Bacterial vaginosis and HIV acquisition: A meta-analysis of published studies. AIDS 2008, 22, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Buve, A.; Jespers, V.; Crucitti, T.; Fichorova, R.N. The vaginal microbiota and susceptibility to HIV. AIDS 2014, 28, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- Seña, A.C.; Goldstein, L.A.; Ramirez, G.; Parish, A.J.; McClelland, R.S. Bacterial Vaginosis and Its Association With Incident Trichomonas vaginalis Infections: A Systematic Review and Meta-Analysis. Sex. Transm. Dis. 2021, 48, e192–e201. [Google Scholar] [CrossRef]
- Rowley, J.; Vander Hoorn, S.; Korenromp, E.; Low, N.; Unemo, M.; Abu-Raddad, L.J.; Chico, R.M.; Smolak, A.; Newman, L.; Gottlieb, S.; et al. Chlamydia, gonorrhoea, trichomoniasis and syphilis: Global prevalence and incidence estimates, 2016. Bull. World Health Organ. 2019, 97, 548–562. [Google Scholar] [CrossRef]
- Fichorova, R.N. Impact of T. vaginalis infection on innate immune responses and reproductive outcome. J. Reprod. Immunol. 2009, 83, 185–189. [Google Scholar] [CrossRef]
- Silver, B.J.; Guy, R.J.; Kaldor, J.M.; Jamil, M.S.; Rumbold, A.R. Trichomonas vaginalis as a Cause of Perinatal Morbidity: A Systematic Review and Meta-Analysis. Sex. Transm. Dis. 2014, 41, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Van Gerwen, O.; Craig-Kuhn, M.; Jones, A.; Schroeder, J.; Deaver, J.; Buekens, P.; Kissinger, P.; Muzny, C. Trichomoniasis and adverse birth outcomes: A systematic review and meta-analysis. BJOG Int. J. Obstet. Gynaecol. 2021, 128, 1907–1915. [Google Scholar] [CrossRef] [PubMed]
- Gülmezoglu, A.M.; Azhar, M. Interventions for trichomoniasis in pregnancy. Cochrane Database Syst. Rev. 2011, 2011, CD000220. [Google Scholar] [CrossRef] [PubMed]
- Fichorova, R.N.; Lee, Y.; Yamamoto, H.S.; Takagi, Y.; Hayes, G.R.; Goodman, R.P.; Chepa-Lotrea, X.; Buck, O.R.; Murray, R.; Kula, T.; et al. Endobiont viruses sensed by the human host—Beyond conventional antiparasitic therapy. PLoS ONE 2012, 7, e48418. [Google Scholar] [CrossRef]
- Hinderfeld, A.S.; Simoes-Barbosa, A. Vaginal dysbiotic bacteria act as pathobionts of the protozoal pathogen Trichomonas vaginalis. Microb. Pathog. 2020, 138, 103820. [Google Scholar] [CrossRef]
- Brotman, R.M.; Bradford, L.L.; Conrad, M.; Gajer, P.; Ault, K.; Peralta, L.; Forney, L.J.; Carlton, J.M.; Abdo, Z.; Ravel, J. Association between Trichomonas vaginalis and vaginal bacterial community composition among reproductive-age women. Sex. Transm. Dis 2012, 39, 807–812. [Google Scholar] [CrossRef]
- Engels, B.M.; Hutvagner, G. Principles and effects of microRNA-mediated post-transcriptional gene regulation. Oncogene 2006, 25, 6163–6169. [Google Scholar] [CrossRef]
- Lai, E.C. Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat. Genet. 2002, 30, 363–364. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Woith, E.; Fuhrmann, G.; Melzig, M.F. Extracellular Vesicles—Connecting Kingdoms. Int. J. Mol. Sci. 2019, 20, 5695. [Google Scholar] [CrossRef]
- Marcilla, A.; Martin-Jaular, L.; Trelis, M.; de Menezes-Neto, A.; Osuna, A.; Bernal, D.; Fernandez-Becerra, C.; Almeida, I.C.; Del Portillo, H.A. Extracellular vesicles in parasitic diseases. J. Extracell. Vesicles 2014, 3, 25040. [Google Scholar] [CrossRef]
- Twu, O.; de Miguel, N.; Lustig, G.; Stevens, G.C.; Vashisht, A.A.; Wohlschlegel, J.A.; Johnson, P.J. Trichomonas vaginalis exosomes deliver cargo to host cells and mediate host:parasite interactions. PLoS Pathog. 2013, 9, e1003482. [Google Scholar] [CrossRef]
- Nievas, Y.R.; Coceres, V.M.; Midlej, V.; de Souza, W.; Benchimol, M.; Pereira-Neves, A.; Vashisht, A.A.; Wohlschlegel, J.A.; Johnson, P.J.; de Miguel, N. Membrane-shed vesicles from the parasite Trichomonas vaginalis: Characterization and their association with cell interaction. Cell Mol. Life Sci. 2018, 75, 2211–2226. [Google Scholar] [CrossRef]
- Rai, A.K.; Johnson, P.J. Trichomonas vaginalis extracellular vesicles are internalized by host cells using proteoglycans and caveolin-dependent endocytosis. Proc. Natl. Acad. Sci. USA 2019, 116, 21354–21360. [Google Scholar] [CrossRef]
- Govender, Y.; Chan, T.; Yamamoto, H.S.; Budnik, B.; Fichorova, R.N. The Role of Small Extracellular Vesicles in Viral-Protozoan Symbiosis: Lessons From Trichomonasvirus in an Isogenic Host Parasite Model. Front. Cell Infect. Microbiol. 2020, 10, 591172. [Google Scholar] [CrossRef]
- Fichorova, R.N.; Rheinwald, J.G.; Anderson, D.J. Generation of papillomavirus-immortalized cell lines from normal human ectocervical, endocervical, and vaginal epithelium that maintain expression of tissue-specific differentiation proteins. Biol. Reprod. 1997, 57, 847–855. [Google Scholar] [CrossRef]
- Fichorova, R.N.; Yamamoto, H.S.; Delaney, M.L.; Onderdonk, A.B.; Doncel, G.F. Novel vaginal microflora colonization model providing new insight into microbicide mechanism of action. mBio 2011, 2, e00168-00111. [Google Scholar] [CrossRef]
- Fichorova, R.N.; Trifonova, R.T.; Gilbert, R.O.; Costello, C.E.; Hayes, G.R.; Lucas, J.J.; Singh, B.N. Trichomonas vaginalis lipophosphoglycan triggers a selective upregulation of cytokines by human female reproductive tract epithelial cells. Infect. Immun. 2006, 74, 5773–5779. [Google Scholar] [CrossRef] [PubMed]
- Fichorova, R.N.; Yamamoto, H.S.; Fashemi, T.; Foley, E.; Ryan, S.; Beatty, N.; Dawood, H.; Hayes, G.R.; St-Pierre, G.; Sato, S.; et al. Trichomonas vaginalis Lipophosphoglycan Exploits Binding to Galectin-1 and -3 to Modulate Epithelial Immunity. J. Biol. Chem. 2016, 291, 998–1013. [Google Scholar] [CrossRef] [PubMed]
- Heiss, C.; Wang, Z.; Black, I.; Azadi, P.; Fichorova, R.N.; Singh, B.N. Novel structural features of the immunocompetent ceramide phospho-inositol glycan core from Trichomonas vaginalis. Carbohydr. Res. 2016, 419, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Hayes, G.R.; Lucas, J.J.; Sommer, U.; Viseux, N.; Mirgorodskaya, E.; Trifonova, R.T.; Sassi, R.R.; Costello, C.E.; Fichorova, R.N. Structural details and composition of Trichomonas vaginalis lipophosphoglycan in relevance to the epithelial immune function. Glycoconj. J. 2009, 26, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Fichorova, R.N.; Buck, O.R.; Yamamoto, H.S.; Fashemi, T.; Dawood, H.Y.; Fashemi, B.; Hayes, G.R.; Beach, D.H.; Takagi, Y.; Delaney, M.L.; et al. The villain team-up or how Trichomonas vaginalis and bacterial vaginosis alter innate immunity in concert. Sex. Transm. Infect. 2013, 89, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Shelly, C.E.; Filatava, E.J.; Thai, J.; Pados, B.F.; Rostas, S.E.; Yamamoto, H.; Fichorova, R.; Gregory, K.E. Elevated Intestinal Inflammation in Preterm Infants With Signs and Symptoms of Gastroesophageal Reflux Disease. Biol. Res. Nurs. 2021, 23, 524–532. [Google Scholar] [CrossRef]
- Goodman, R.P.; Freret, T.S.; Kula, T.; Geller, A.M.; Talkington, M.W.; Tang-Fernandez, V.; Suciu, O.; Demidenko, A.A.; Ghabrial, S.A.; Beach, D.H.; et al. Clinical isolates of Trichomonas vaginalis concurrently infected by strains of up to four Trichomonasvirus species (Family Totiviridae). J. Virol. 2011, 85, 4258–4270. [Google Scholar] [CrossRef]
- Goodman, R.P.; Ghabrial, S.A.; Fichorova, R.N.; Nibert, M.L. Trichomonasvirus: A new genus of protozoan viruses in the family Totiviridae. Arch. Virol. 2011, 156, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Parent, K.N.; Takagi, Y.; Cardone, G.; Olson, N.H.; Ericsson, M.; Yang, M.; Lee, Y.; Asara, J.M.; Fichorova, R.N.; Baker, T.S.; et al. Structure of a protozoan virus from the human genitourinary parasite Trichomonas vaginalis. mBio 2013, 4, e00056-13. [Google Scholar] [CrossRef]
- Onderdonk, A.B.; Zamarchi, G.R.; Rodriguez, M.L.; Hirsch, M.L.; Munoz, A.; Kass, E.H. Qualitative assessment of vaginal microflora during use of tampons of various compositions. Appl. Environ. Microbiol. 1987, 53, 2779–2784. [Google Scholar] [CrossRef]
- Fashemi, B.; Delaney, M.L.; Onderdonk, A.B.; Fichorova, R.N. Effects of feminine hygiene products on the vaginal mucosal biome. Microb. Ecol. Health Dis. 2013, 24, 19703. [Google Scholar] [CrossRef]
- Fichorova, R.N.; DeLong, A.K.; Cu-Uvin, S.; King, C.C.; Jamieson, D.J.; Klein, R.S.; Sobel, J.D.; Vlahov, D.; Yamamoto, H.S.; Mayer, K.H. Protozoan-Viral-Bacterial Co-Infections Alter Galectin Levels and Associated Immunity Mediators in the Female Genital Tract. Front. Cell Infect. Microbiol. 2021, 11, 649940. [Google Scholar] [CrossRef]
- Foy, J.P.; Bertolus, C.; Michallet, M.C.; Deneuve, S.; Incitti, R.; Bendriss-Vermare, N.; Albaret, M.A.; Ortiz-Cuaran, S.; Thomas, E.; Colombe, A.; et al. The immune microenvironment of HPV-negative oral squamous cell carcinoma from never-smokers and never-drinkers patients suggests higher clinical benefit of IDO1 and PD1/PD-L1 blockade. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 1934–1941. [Google Scholar] [CrossRef]
- Girard, L.; Rodriguez-Canales, J.; Behrens, C.; Thompson, D.M.; Botros, I.W.; Tang, H.; Xie, Y.; Rekhtman, N.; Travis, W.D.; Wistuba, I.I.; et al. An Expression Signature as an Aid to the Histologic Classification of Non-Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4880–4889. [Google Scholar] [CrossRef] [PubMed]
- Lizarraga, D.; Huen, K.; Combs, M.; Escudero-Fung, M.; Eskenazi, B.; Holland, N. miRNAs differentially expressed by next-generation sequencing in cord blood buffy coat samples of boys and girls. Epigenomics 2016, 8, 1619–1635. [Google Scholar] [CrossRef] [PubMed]
- Reed, E.R.; Latourelle, J.C.; Bockholt, J.H.; Bregu, J.; Smock, J.; Paulsen, J.S.; Myers, R.H. MicroRNAs in CSF as prodromal biomarkers for Huntington disease in the PREDICT-HD study. Neurology 2018, 90, e264–e272. [Google Scholar] [CrossRef] [PubMed]
- Satake, E.; Pezzolesi, M.G.; Md Dom, Z.I.; Smiles, A.M.; Niewczas, M.A.; Krolewski, A.S. Circulating miRNA Profiles Associated With Hyperglycemia in Patients With Type 1 Diabetes. Diabetes 2018, 67, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Retz, M.; Siefker-Radtke, A.; Baron, A.; Necchi, A.; Bedke, J.; Plimack, E.R.; Vaena, D.; Grimm, M.O.; Bracarda, S.; et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017, 18, 312–322. [Google Scholar] [CrossRef]
- Shlomi, T.; Herrgard, M.; Portnoy, V.; Naim, E.; Palsson, B.; Sharan, R.; Ruppin, E. Systematic condition-dependent annotation of metabolic genes. Genome Res. 2007, 17, 1626–1633. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2020, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Korcsmáros, T.; Kiss, H.J.; London, G.; Nussinov, R. Structure and dynamics of molecular networks: A novel paradigm of drug discovery: A comprehensive review. Pharm. Ther. 2013, 138, 333–408. [Google Scholar] [CrossRef]
- Willms, E.; Cabañas, C.; Mäger, I.; Wood, M.J.A.; Vader, P. Extracellular Vesicle Heterogeneity: Subpopulations, Isolation Techniques, and Diverse Functions in Cancer Progression. Front. Immunol. 2018, 9, 738. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, Y.; Xiong, X.; Chen, J.; Tang, W.; He, L.; Zhang, Z.; Yin, Y.; Li, F. Intestinal accumulation of microbiota-produced succinate caused by loss of microRNAs leads to diarrhea in weanling piglets. Gut Microbes 2022, 14, 2091369. [Google Scholar] [CrossRef]
- Fichorova, R.; Fraga, J.; Rappelli, P.; Fiori, P.L. Trichomonas vaginalis infection in symbiosis with Trichomonasvirus and Mycoplasma. Res. Microbiol. 2017, 168, 882–891. [Google Scholar] [CrossRef]
- Gajer, P.; Brotman, R.M.; Bai, G.; Sakamoto, J.; Schutte, U.M.; Zhong, X.; Koenig, S.S.; Fu, L.; Ma, Z.S.; Zhou, X.; et al. Temporal dynamics of the human vaginal microbiota. Sci. Transl. Med. 2012, 4, 132ra152. [Google Scholar] [CrossRef]
- Nahui Palomino, R.A.; Vanpouille, C.; Laghi, L.; Parolin, C.; Melikov, K.; Backlund, P.; Vitali, B.; Margolis, L. Extracellular vesicles from symbiotic vaginal lactobacilli inhibit HIV-1 infection of human tissues. Nat. Commun. 2019, 10, 5656. [Google Scholar] [CrossRef]
- Cheng, L.; Kazmierczak, D.; Norenhag, J.; Hamsten, M.; Fransson, E.; Schuppe-Koistinen, I.; Olovsson, M.; Engstrand, L.; Hydbring, P.; Du, J. A MicroRNA Gene Panel Predicts the Vaginal Microbiota Composition. mSystems 2021, 6, e00175-21. [Google Scholar] [CrossRef]
- Bartel, D.P.; Chen, C.-Z. Micromanagers of gene expression: The potentially widespread influence of metazoan microRNAs. Nat. Rev. Genet. 2004, 5, 396–400. [Google Scholar] [CrossRef]
- Liu, J.; Sun, H.; Wang, X.; Yu, Q.; Li, S.; Yu, X.; Gong, W. Increased Exosomal MicroRNA-21 and MicroRNA-146a Levels in the Cervicovaginal Lavage Specimens of Patients with Cervical Cancer. Int. J. Mol. Sci. 2014, 15, 758–773. [Google Scholar] [CrossRef] [PubMed]
- Azimi, T.; Paryan, M.; Mondanizadeh, M.; Sarmadian, H.; Zamani, A. Pap Smear miR-92a-5p and miR-155-5p as potential diagnostic biomarkers of squamous intraepithelial cervical cancer. Asian Pac. J. Cancer Prev. 2021, 22, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- de Castro-Sobrinho, J.M.; Rabelo-Santos, S.H.; Fugueiredo-Alves, R.R.; Derchain, S.; Sarian, L.O.; Pitta, D.R.; Campos, E.A.; Zeferino, L.C. Bacterial vaginosis and inflammatory response showed association with severity of cervical neoplasia in HPV-positive women. Diagn. Cytopathol. 2016, 44, 80–86. [Google Scholar] [CrossRef]
- Romero-Morelos, P.; Bandala, C.; Jiménez-Tenorio, J.; Valdespino-Zavala, M.; Rodríguez-Esquivel, M.; Gama-Ríos, R.A.; Bandera, A.; Mendoza-Rodríguez, M.; Taniguchi, K.; Marrero-Rodríguez, D.; et al. Vaginosis-associated bacteria and its association with HPV infection. Med. Clin. 2019, 152, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A. Protein Interaction Networks: Computational Analysis; Cambridge University Press: Cambridge, MA, USA, 2009. [Google Scholar]
- Villa, P.; Cipolla, C.; D’Ippolito, S.; Amar, I.D.; Shachor, M.; Ingravalle, F.; Scaldaferri, F.; Puca, P.; Di Simone, N.; Scambia, G. The interplay between immune system and microbiota in gynecological diseases: A narrative review. Eur. Rev. Med. Pharm. Sci. 2020, 24, 5676–5690. [Google Scholar] [CrossRef]
- Capece, A.; Vasieva, O.; Meher, S.; Alfirevic, Z.; Alfirevic, A. Pathway Analysis of Genetic Factors Associated with Spontaneous Preterm Birth and Pre-Labor Preterm Rupture of Membranes. PLoS ONE 2014, 9, e108578. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, S.; Seddighzadeh, B.; Baccarelli, A.; Wise, L.A.; Williams, M.; Shields, A.E. Adverse maternal exposures, methylation of glucocorticoid-related genes and perinatal outcomes: A systematic review. Epigenomics 2016, 8, 925–944. [Google Scholar] [CrossRef]
- Chalfun, G.; Reis, M.M.; de Oliveira, M.B.G.; de Araújo Brasil, A.; Dos Santos Salú, M.; da Cunha, A.; Prata-Barbosa, A.; de Magalhães-Barbosa, M.C. Perinatal stress and methylation of the NR3C1 gene in newborns: Systematic review. Epigenetics 2022, 17, 1003–1019. [Google Scholar] [CrossRef]
- Chen, P.; Li, B.; Ou-Yang, L. Role of estrogen receptors in health and disease. Front. Endocrinol. 2022, 13, 839005. [Google Scholar] [CrossRef]
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol. 2015, 294, 63–69. [Google Scholar] [CrossRef]
- Shivers, K.Y.; Amador, N.; Abrams, L.; Hunter, D.; Jenab, S.; Quiñones-Jenab, V. Estrogen alters baseline and inflammatory-induced cytokine levels independent from hypothalamic-pituitary-adrenal axis activity. Cytokine 2015, 72, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.Z.; Cidlowski, J.A. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol. Cell 2005, 18, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Nazzari, S.; Grumi, S.; Mambretti, F.; Villa, M.; Giorda, R.; Provenzi, L. Maternal and infant NR3C1 and SLC6A4 epigenetic signatures of the COVID-19 pandemic lockdown: When timing matters. Transl. Psychiatry 2022, 12, 386. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, Z.; Vlaikou, A.-M.; Theodoridou, D.; Markopoulos, G.S.; Tsoni, K.; Agakidou, E.; Drosou-Agakidou, V.; Turck, C.W.; Filiou, M.D.; Syrrou, M. Stressful Newborn Memories: Pre-Conceptual, In Utero, and Postnatal Events. Front. Psychiatry 2019, 10, 220. [Google Scholar] [CrossRef]
- Forbes, K.; Westwood, M. The IGF axis and placental function. a mini review. Horm. Res. 2008, 69, 129–137. [Google Scholar] [CrossRef]
- Solano, M.E.; Arck, P.C. Steroids, Pregnancy and Fetal Development. Front. Immunol. 2020, 10, 3017. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 145. [Google Scholar] [CrossRef]
- Lv, A.; Tu, Z.; Huang, Y.; Lu, W.; Xie, B. Circulating exosomal miR-125a-5p as a novel biomarker for cervical cancer. Oncol. Lett. 2021, 21, 54. [Google Scholar] [CrossRef]
- Royston, S.E.; Yasui, N.; Kondilis, A.G.; Lord, S.V.; Katzenellenbogen, J.A.; Mahoney, M.M. ESR1 and ESR2 differentially regulate daily and circadian activity rhythms in female mice. Endocrinology 2014, 155, 2613–2623. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Chang, A.K.; Zang, M.X.; Bi, H.; Li, S.; Wang, M.; Xing, X.; Wu, H. Induction of the CLOCK gene by E2-ERα signaling promotes the proliferation of breast cancer cells. PLoS ONE 2014, 9, e95878. [Google Scholar] [CrossRef]
- Roberts, C.W.; Walker, W.; Alexander, J. Sex-associated hormones and immunity to protozoan parasites. Clin. Microbiol. Rev. 2001, 14, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hub Rank | Gene Symbol | Gene Name | Centrality Score * | Degree ** | EV-miRNAs Targeting Hub Genes | Number of 3′UTR Target Sites |
|---|---|---|---|---|---|---|
| 1 | MAPK1 | mitogen-activated protein kinase 1 | 26,313.7 | 53 | hsa-miR-106b-5p, hsa-miR-20a-5p, hsa-miR-6088 | 4 |
| 2 | MAPK14 | mitogen-activated protein kinase 14 | 25,464.1 | 40 | hsa-miR-24-3p | 1 |
| 3 | ESR1 | estrogen receptor 1 | 24,999 | 34 | hsa-miR-130a-3p, hsa-miR-130b-3p, hsa-miR-18a-5p, hsa-miR-20b-5p, hsa-miR-22-3p, hsa-miR-221-3p, hsa-miR-222-3p, hsa-miR-454-3p | 22 |
| 4 | SMAD4 | SMAD family member 4 | 22,813.3 | 41 | hsa-miR-449a | 2 |
| 5 | PIK3R1 | phosphoinositide-3-kinase regulatory subunit 1 | 19,971.2 | 46 | hsa-miR-103a-3p, hsa-miR-107, hsa-miR-128-3p | 11 |
| 6 | UBE2I | Ubiquitin Conjugating Enzyme E2 I | 19,821.3 | 32 | hsa-miR-188-5p | 3 |
| 7 | NR3C1 | nuclear receptor subfamily 3 group C member 1 | 19,570.7 | 24 | hsa-miR-18a-5p | 4 |
| 8 | STAT3 | signal transducer and activator of transcription 3 | 17,914.2 | 39 | hsa-miR-125a-5p | 18 |
| 9 | CDC42 | cell division cycle 42 | 16,943.4 | 34 | hsa-miR-185-5p | 3 |
| 10 | YWHAZ | tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein zeta | 15,952.3 | 29 | hsa-miR-193a-3p, hsa-miR-193b-3p | 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cezar-de-Mello, P.F.T.; Ryan, S.; Fichorova, R.N. The microRNA Cargo of Human Vaginal Extracellular Vesicles Differentiates Parasitic and Pathobiont Infections from Colonization by Homeostatic Bacteria. Microorganisms 2023, 11, 551. https://doi.org/10.3390/microorganisms11030551
Cezar-de-Mello PFT, Ryan S, Fichorova RN. The microRNA Cargo of Human Vaginal Extracellular Vesicles Differentiates Parasitic and Pathobiont Infections from Colonization by Homeostatic Bacteria. Microorganisms. 2023; 11(3):551. https://doi.org/10.3390/microorganisms11030551
Chicago/Turabian StyleCezar-de-Mello, Paula Fernandes Tavares, Stanthia Ryan, and Raina N. Fichorova. 2023. "The microRNA Cargo of Human Vaginal Extracellular Vesicles Differentiates Parasitic and Pathobiont Infections from Colonization by Homeostatic Bacteria" Microorganisms 11, no. 3: 551. https://doi.org/10.3390/microorganisms11030551
APA StyleCezar-de-Mello, P. F. T., Ryan, S., & Fichorova, R. N. (2023). The microRNA Cargo of Human Vaginal Extracellular Vesicles Differentiates Parasitic and Pathobiont Infections from Colonization by Homeostatic Bacteria. Microorganisms, 11(3), 551. https://doi.org/10.3390/microorganisms11030551