
Avian Malaria Parasites Modulate Gut Microbiome Assembly in Canaries
 , , , ,
, , , ,  , and
, and 
Abstract
:1. Introduction
2. Material and Methods
2.1. Ethical Statement
2.2. Birds and Housing Conditions
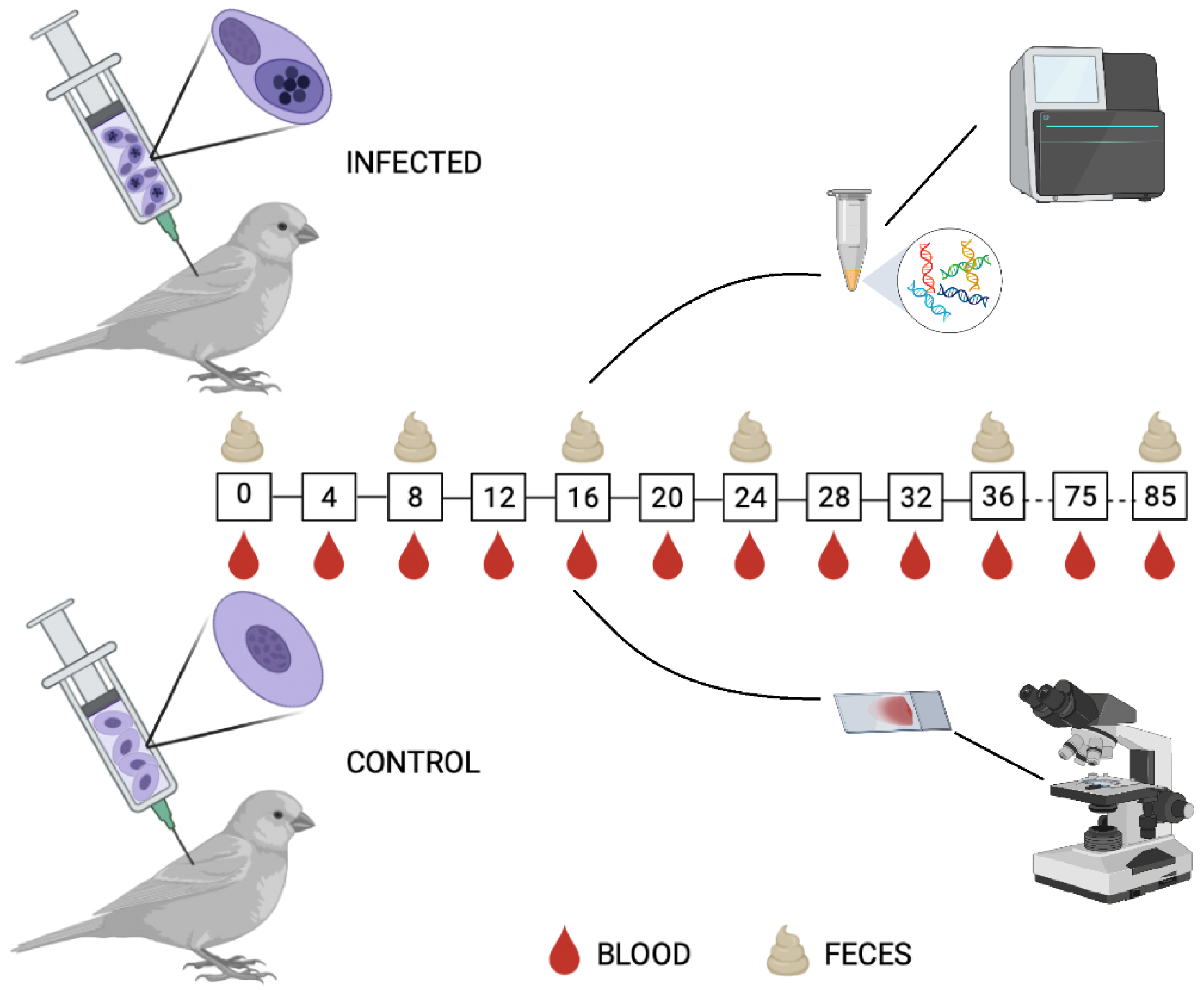
2.3. Experimental Design
2.4. Thawing the Cryopreserved Avian Malaria Sample
2.5. Experimental Infection of Birds
2.6. Blood Sample and Bird Feces Collection
2.7. Microscopic Examination
2.8. DNA Extraction and PCR for P. homocircumflexum Identification
2.9. DNA Extraction and 16S rRNA Sequencing for Microbiome Analysis
2.10. 16S rRNA Sequences Processing
2.11. Statistical Analysis
2.12. Bacterial Co-Occurrence Networks and Attack Tolerance Test
2.13. Prediction of Functional Traits in the Bird Microbiome
3. Results
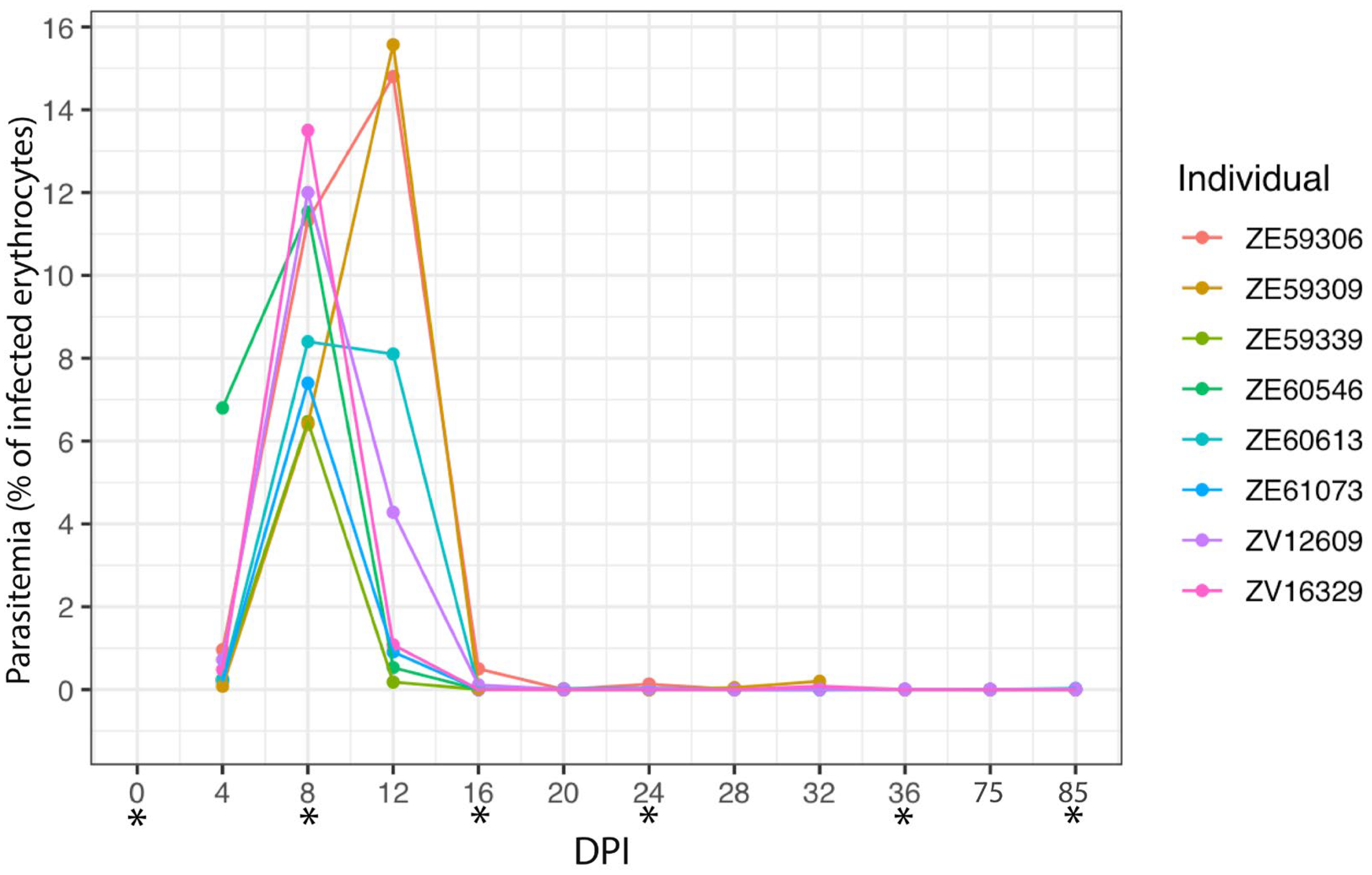
3.1. Experimental Infection of Plasmodium homocircumflexum in Birds
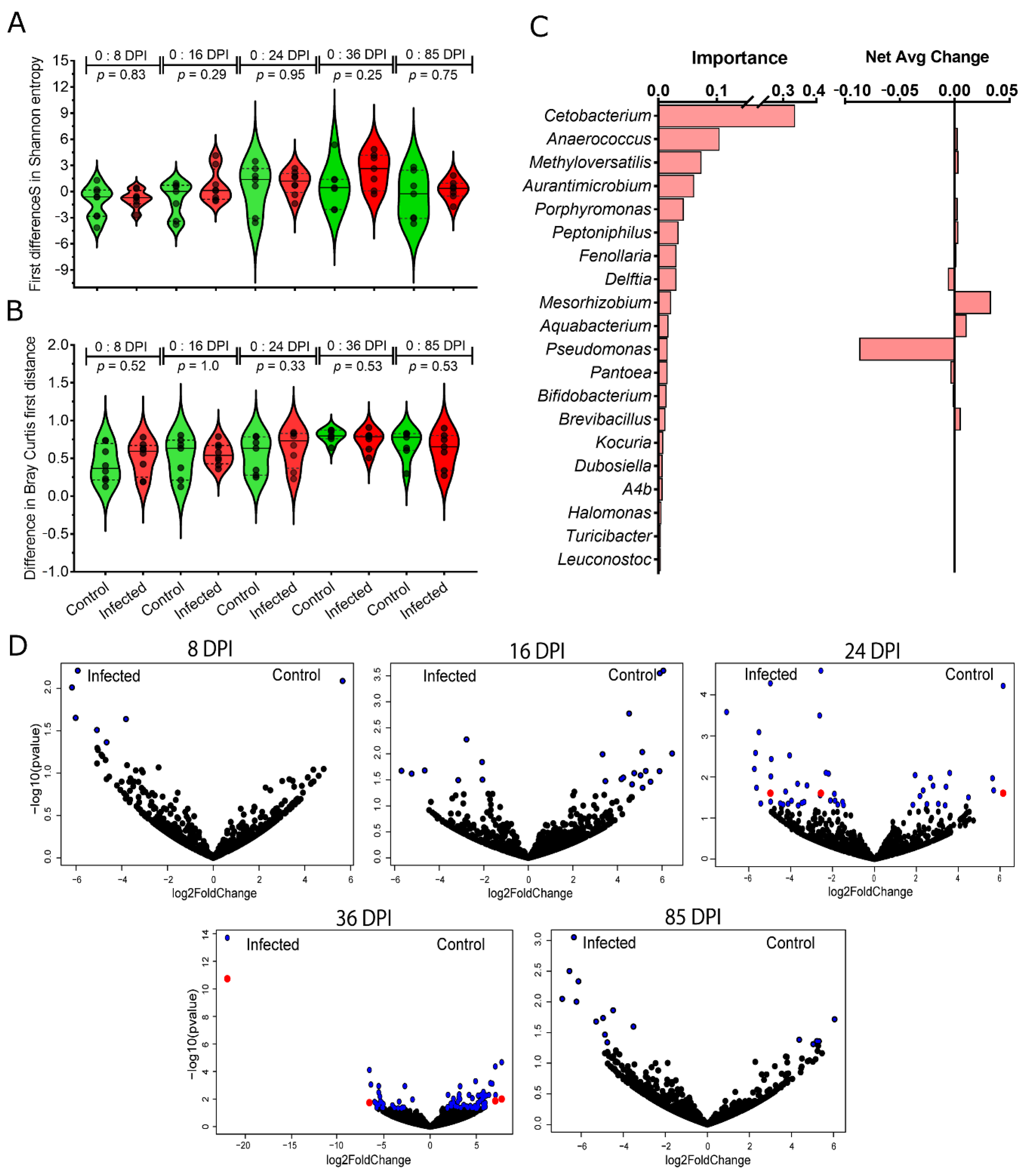
3.2. Changes in the Taxonomic Profiles of Bird Gut Microbiome in Response to P. homocircumflexum Infection
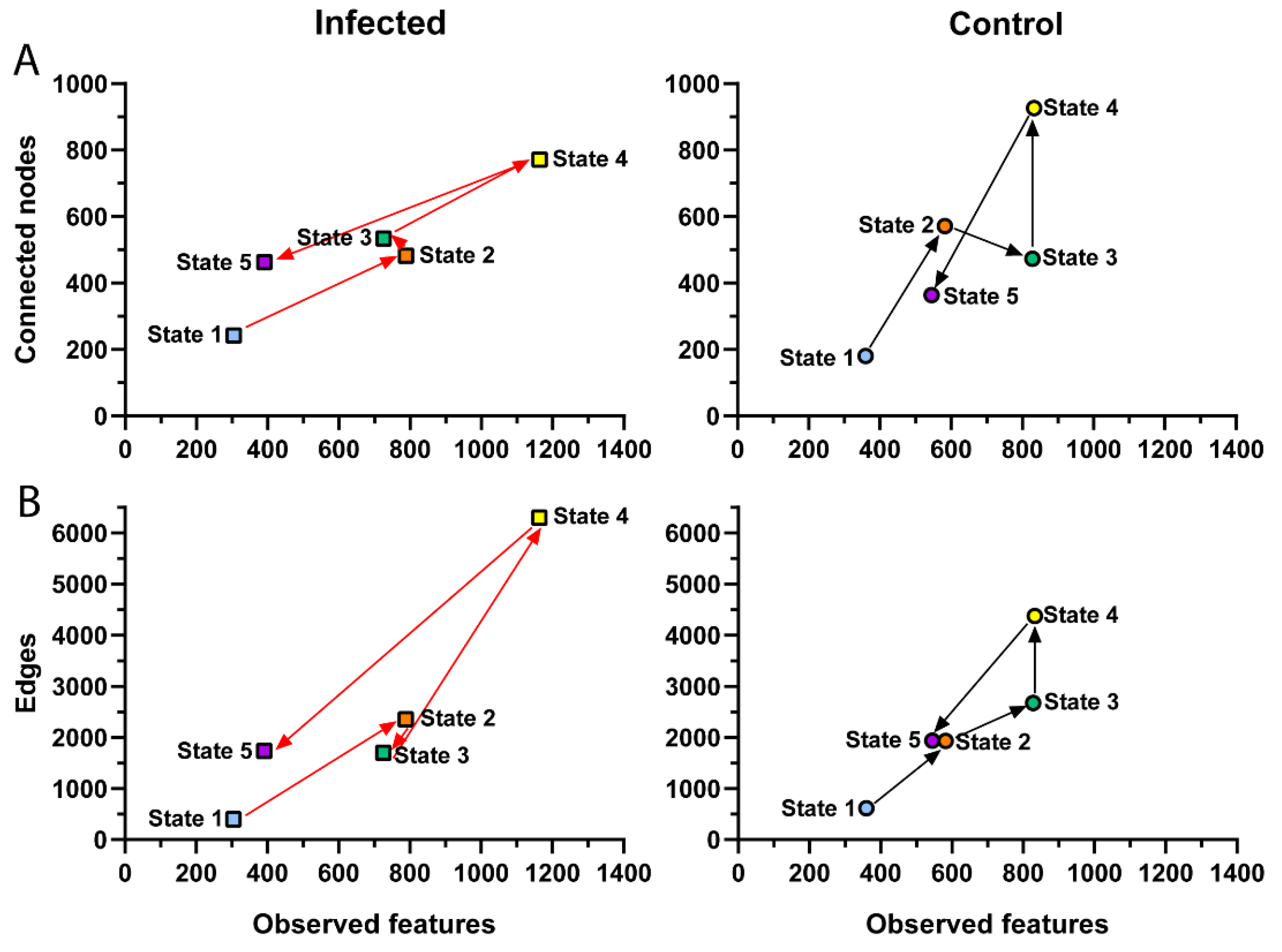
3.3. Changes in Microbial Community Assembly in Response to P. homocircumflexum Infection
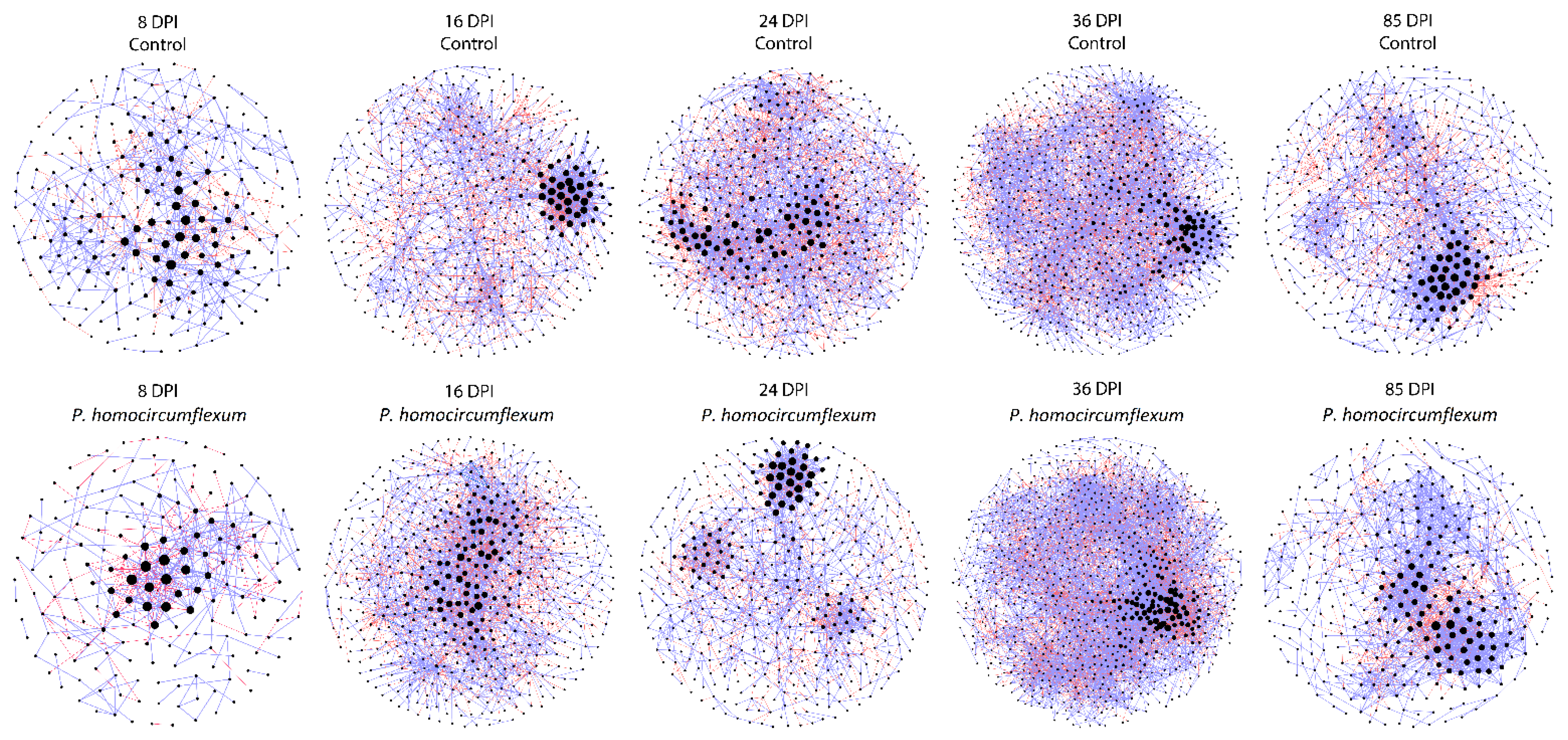
3.4. Impact of P. homocircumflexum Infection on Network Centrality Distribution and Robustness
3.5. Impact of P. homocircumflexum Infection on the Functional Profiles of Bird Guts Microbiome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grond, K.; Sandercock, B.K.; Jumpponen, A.; Zeglin, L.H. The Avian Gut Microbiota: Community, Physiology and Function in Wild Birds. J. Avian Biol. 2018, 49, e01788. [Google Scholar] [CrossRef] [Green Version]
- Hird, S.M.; Sánchez, C.; Carstens, B.C.; Brumfield, R.T. Comparative Gut Microbiota of 59 Neotropical Bird Species. Front. Microbiol. 2015, 6, 1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodawatta, K.H.; Hird, S.M.; Grond, K.; Poulsen, M.; Jønsson, K.A. Avian Gut Microbiomes Taking Flight. Trends Microbiol. 2022, 30, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Waite, D.W.; Taylor, M.W. Characterizing the Avian Gut Microbiota: Membership, Driving Influences, and Potential Function. Front. Microbiol. 2014, 5, 223. [Google Scholar] [CrossRef] [Green Version]
- Grond, K.; Santo Domingo, J.W.; Lanctot, R.B.; Jumpponen, A.; Bentzen, R.L.; Boldenow, M.L.; Brown, S.C.; Casler, B.; Cunningham, J.A.; Doll, A.C.; et al. Composition and Drivers of Gut Microbial Communities in Arctic-Breeding Shorebirds. Front. Microbiol. 2019, 10, 2258. [Google Scholar] [CrossRef] [Green Version]
- Hird, S.M.; Carstens, B.C.; Cardiff, S.W.; Dittmann, D.L.; Brumfield, R.T. Sampling Locality Is More Detectable than Taxonomy or Ecology in the Gut Microbiota of the Brood-Parasitic Brown-Headed Cowbird (Molothrus ater). PeerJ 2014, 2, e321. [Google Scholar] [CrossRef] [Green Version]
- Palinauskas, V.; Mateos-Hernandez, L.; Wu-Chuang, A.; de la Fuente, J.; Aželytė, J.; Obregon, D.; Cabezas-Cruz, A. Exploring the Ecological Implications of Microbiota Diversity in Birds: Natural Barriers Against Avian Malaria. Front. Immunol. 2022, 13, 807682. [Google Scholar] [CrossRef]
- Mammeri, M.; Obregón, D.A.; Chevillot, A.; Polack, B.; Julien, C.; Pollet, T.; Cabezas-Cruz, A.; Adjou, K.T. Cryptosporidium Parvum Infection Depletes Butyrate Producer Bacteria in Goat Kid Microbiome. Front. Microbiol. 2020, 11, 548737. [Google Scholar] [CrossRef]
- Madlala, T.; Okpeku, M.; Adeleke, M.A. Understanding the Interactions between Eimeria Infection and Gut Microbiota, towards the Control of Chicken Coccidiosis: A Review. Parasite 2021, 28, 48. [Google Scholar] [CrossRef]
- Videvall, E.; Paxton, K.L.; Campana, M.G.; Cassin-Sackett, L.; Atkinson, C.T.; Fleischer, R.C. Transcriptome Assembly and Differential Gene Expression of the Invasive Avian Malaria Parasite Plasmodium Relictum in Hawaiʻi. Ecol. Evol. 2021, 11, 4935–4944. [Google Scholar] [CrossRef]
- Videvall, E.; Marzal, A.; Magallanes, S.; Fleischer, R.C.; Espinoza, K.; García-Longoria, L. The Uropygial Gland Microbiome of House Sparrows with Malaria Infection. J. Avian Biol. 2021, 52, jav.02686. [Google Scholar] [CrossRef]
- Bamgbose, T.; Anvikar, A.R.; Alberdi, P.; Abdullahi, I.O.; Inabo, H.I.; Bello, M.; Cabezas-Cruz, A.; de la Fuente, J. Functional Food for the Stimulation of the Immune System Against Malaria. Probiotics Antimicro. Prot. 2021, 13, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, M.M.; Denny, J.E.; Langelier, C.; Sears, C.L.; Schmidt, N.W. Malaria and the Microbiome: A Systematic Review. Clin. Infect. Dis. 2018, 67, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, D.; Chora, Â.F.; Mota, M.M. Microbiota, a Third Player in the Host–Plasmodium Affair. Trends Parasitol. 2020, 36, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Mooney, J.P.; Lokken, K.L.; Byndloss, M.X.; George, M.D.; Velazquez, E.M.; Faber, F.; Butler, B.P.; Walker, G.T.; Ali, M.M.; Potts, R.; et al. Inflammation-Associated Alterations to the Intestinal Microbiota Reduce Colonization Resistance against Non-Typhoidal Salmonella during Concurrent Malaria Parasite Infection. Sci. Rep. 2015, 5, 14603. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, T.; Miyauchi, E.; Nakamura, S.; Hirai, M.; Suzue, K.; Imai, T.; Nomura, T.; Handa, T.; Okada, H.; Shimokawa, C.; et al. Plasmodium Berghei ANKA Causes Intestinal Malaria Associated with Dysbiosis. Sci. Rep. 2015, 5, 15699. [Google Scholar] [CrossRef] [Green Version]
- Valkiūnas, G.; Iezhova, T.A. Keys to the Avian Malaria Parasites. Malar. J. 2018, 17, 212. [Google Scholar] [CrossRef] [Green Version]
- Bensch, S.; Hellgren, O.; Pérez-Tris, J. MalAvi: A Public Database of Malaria Parasites and Related Haemosporidians in Avian Hosts Based on Mitochondrial Cytochrome b Lineages. Mol. Ecol. Resour. 2009, 9, 1353–1358. [Google Scholar] [CrossRef]
- Valkiūnas, G. Avian Malaria Parasites and Other Haemosporidia; CRC Press: Boca Raton, FL, USA, 2005; ISBN 978-0-415-30097-1. [Google Scholar]
- Palinauskas, V.; Žiegytė, R.; Ilgūnas, M.; Iezhova, T.A.; Bernotienė, R.; Bolshakov, C.; Valkiūnas, G. Description of the First Cryptic Avian Malaria Parasite, Plasmodium Homocircumflexum n. Sp., with Experimental Data on Its Virulence and Development in Avian Hosts and Mosquitoes. Int. J. Parasitol. 2015, 45, 51–62. [Google Scholar] [CrossRef]
- Ilgūnas, M.; Bukauskaitė, D.; Palinauskas, V.; Iezhova, T.A.; Dinhopl, N.; Nedorost, N.; Weissenbacher-Lang, C.; Weissenböck, H.; Valkiūnas, G. Mortality and Pathology in Birds due to Plasmodium (Giovannolaia) Homocircumflexum Infection, with Emphasis on the Exoerythrocytic Development of Avian Malaria Parasites. Malar. J. 2016, 15, 256. [Google Scholar] [CrossRef] [Green Version]
- Ilgūnas, M.; Bukauskaitė, D.; Palinauskas, V.; Iezhova, T.; Fragner, K.; Platonova, E.; Weissenböck, H.; Valkiūnas, G. Patterns of Plasmodium Homocircumflexum Virulence in Experimentally Infected Passerine Birds. Malar. J. 2019, 18, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Longoria, L.; Palinauskas, V.; Ilgūnas, M.; Valkiūnas, G.; Hellgren, O. Differential Gene Expression of Plasmodium Homocircumflexum (Lineage PCOLL4) across Two Experimentally Infected Passerine Bird Species. Genomics 2020, 112, 2857–2865. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, D.; Palinauskas, V.; Iezhova, T.A.; Bernotienė, R.; Ilgūnas, M.; Bukauskaitė, D.; Zehtindjiev, P.; Ilieva, M.; Shapoval, A.P.; Bolshakov, C.V.; et al. Plasmodium Spp.: An Experimental Study on Vertebrate Host Susceptibility to Avian Malaria. Exp. Parasitol. 2015, 148, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Palinauskas, V.; Valkiūnas, G.; Bolshakov, C.V.; Bensch, S. Plasmodium Relictum (Lineage P-SGS1): Effects on Experimentally Infected Passerine Birds. Exp. Parasitol. 2008, 120, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Valkiūnas, G.; Iezhova, T.A.; Križanauskienė, A.; Palinauskas, V.; Sehgal, R.N.M.; Bensch, S. A Comparative Analysis of Microscopy and PCR-Based Detection Methods for Blood Parasites. J. Parasitol. 2008, 94, 1395–1401. [Google Scholar] [CrossRef]
- Godfrey, R.D.; Fedynich, A.M.; Pence, D.B. Quantification of hematozoa in blood smears. J. Wildl. Dis. 1987, 23, 558–565. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; ISBN 978-0-87969-577-4. [Google Scholar]
- Hellgren, O.; Waldenström, J.; Bensch, S. A new PCR assay for simultaneous studies of Leucocytozoon, Plasmodium, and Haemoproteus from avian blood. J. Parasitol. 2004, 90, 797–802. [Google Scholar] [CrossRef]
- Bensch, S.; Stjernman, M.; Hasselquist, D.; Örjan, Ö.; Hannson, B.; Westerdahl, H.; Pinheiro, R.T. Host Specificity in Avian Blood Parasites: A Study of Plasmodium and Haemoproteus Mitochondrial DNA Amplified from Birds. Proc. R. Soc. Lond. B 2000, 267, 1583–1589. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing Taxonomic Classification of Marker-Gene Amplicon Sequences with QIIME 2’s Q2-Feature-Classifier Plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.-H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the Classification of Cultured and Uncultured Bacteria and Archaea Using 16S RRNA Gene Sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Dillon, M.R.; Zhang, Y.; Rideout, J.R.; Bolyen, E.; Li, H.; Albert, P.S.; Caporaso, J.G. Q2-Longitudinal: Longitudinal and Paired-Sample Analyses of Microbiome Data. mSystems 2018, 3, e00219-18. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Peschel, S.; Müller, C.L.; von Mutius, E.; Boulesteix, A.-L.; Depner, M. NetCoMi: Network Construction and Comparison for Microbiome Data in R. Brief. Bioinform. 2021, 22, bbaa290. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development for R; RStudio, PBC: Boston, MA, USA, 2020; Available online: http://www.Rstudio.com/ (accessed on 20 February 2023).
- Real, R.; Vargas, J.M. The Probabilistic Basis of Jaccard’s Index of Similarity. Syst. Biol. 1996, 45, 380–385. [Google Scholar] [CrossRef]
- Friedman, J.; Alm, E.J. Inferring Correlation Networks from Genomic Survey Data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef] [Green Version]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. ICWSM 2009, 3, 361–362. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S RRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lhomme, S. Analyse Spatiale de La Structure Des Réseaux Techniques Dans Un Contexte de Risques. Cybergeo 2015. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L. The COG Database: A Tool for Genome-Scale Analysis of Protein Functions and Evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc Database of Metabolic Pathways and Enzymes. Nucleic Acids Res. 2018, 46, D633–D639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yooseph, S.; Kirkness, E.F.; Tran, T.M.; Harkins, D.M.; Jones, M.B.; Torralba, M.G.; O’Connell, E.; Nutman, T.B.; Doumbo, S.; Doumbo, O.K.; et al. Stool Microbiota Composition Is Associated with the Prospective Risk of Plasmodium Falciparum Infection. BMC Genom. 2015, 16, 631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colston, T.J.; Jackson, C.R. Microbiome Evolution along Divergent Branches of the Vertebrate Tree of Life: What Is Known and Unknown. Mol. Ecol. 2016, 25, 3776–3800. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Jia, L.; Wei, S.; Ding, H.; Yang, J.; Wang, H. Effects of Eimeria Tenella Infection on the Barrier Damage and Microbiota Diversity of Chicken Cecum. Poult. Sci. 2020, 99, 1297–1305. [Google Scholar] [CrossRef]
- Macdonald, S.E.; van Diemen, P.M.; Martineau, H.; Stevens, M.P.; Tomley, F.M.; Stabler, R.A.; Blake, D.P. Impact of Eimeria Tenella Coinfection on Campylobacter Jejuni Colonization of the Chicken. Infect. Immun. 2019, 87, e00772-18. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, S.E.; Nolan, M.J.; Harman, K.; Boulton, K.; Hume, D.A.; Tomley, F.M.; Stabler, R.A.; Blake, D.P. Effects of Eimeria Tenella Infection on Chicken Caecal Microbiome Diversity, Exploring Variation Associated with Severity of Pathology. PLoS ONE 2017, 12, e0184890. [Google Scholar] [CrossRef]
- Rohrer, S.D.; Robertson, B.Q.; Chubiz, L.M.; Parker, P.G. Gut Microbiome Composition Associated with Plasmodium Infection in the Eurasian Tree Sparrow. J. Avian Biol. 2023, 2023, e03027. [Google Scholar] [CrossRef]
- Guan, W.; Song, X.; Yang, S.; Zhu, H.; Li, F.; Li, J. Observation of the Gut Microbiota Profile in BALB/c Mice Induced by Plasmodium Yoelii 17XL Infection. Front. Microbiol. 2022, 13, 858897. [Google Scholar] [CrossRef] [PubMed]
- Olsson, L.M.; Boulund, F.; Nilsson, S.; Khan, M.T.; Gummesson, A.; Fagerberg, L.; Engstrand, L.; Perkins, R.; Uhlén, M.; Bergström, G.; et al. Dynamics of the Normal Gut Microbiota: A Longitudinal One-Year Population Study in Sweden. Cell Host Microbe 2022, 30, 726–739.e3. [Google Scholar] [CrossRef] [PubMed]
- Priya, S.; Blekhman, R. Population Dynamics of the Human Gut Microbiome: Change Is the Only Constant. Genome Biol. 2019, 20, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belzer, C.; Gerber, G.K.; Roeselers, G.; Delaney, M.; DuBois, A.; Liu, Q.; Belavusava, V.; Yeliseyev, V.; Houseman, A.; Onderdonk, A.; et al. Dynamics of the Microbiota in Response to Host Infection. PLoS ONE 2014, 9, e95534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebreton, L.; Guillerm-Erckelboudt, A.-Y.; Gazengel, K.; Linglin, J.; Ourry, M.; Glory, P.; Sarniguet, A.; Daval, S.; Manzanares-Dauleux, M.J.; Mougel, C. Temporal Dynamics of Bacterial and Fungal Communities during the Infection of Brassica Rapa Roots by the Protist Plasmodiophora Brassicae. PLoS ONE 2019, 14, e0204195. [Google Scholar] [CrossRef] [Green Version]
- Cui, Z.; Huntley, R.B.; Zeng, Q.; Steven, B. Temporal and Spatial Dynamics in the Apple Flower Microbiome in the Presence of the Phytopathogen Erwinia Amylovora. ISME J 2021, 15, 318–329. [Google Scholar] [CrossRef]
- Li, Y.; Li, T.; Zhao, D.; Wang, Z.; Liao, Y. Different Tillage Practices Change Assembly, Composition, and Co-Occurrence Patterns of Wheat Rhizosphere Diazotrophs. Sci. Total Environ. 2021, 767, 144252. [Google Scholar] [CrossRef]
- De Morais, U.L.; Antunes, L. A Look at the Way We Look at Complex Networks Robustness and Resilience. arXiv 2019, arXiv:1909.06448. [Google Scholar] [CrossRef]
- Albert, R.; Jeong, H.; Barabási, A.-L. Error and Attack Tolerance of Complex Networks. Nature 2000, 406, 378–382. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.M.; Moreira, A.A.; Andrade, J.S.; Havlin, S.; Herrmann, H.J. Mitigation of Malicious Attacks on Networks. Proc. Natl. Acad. Sci. USA 2011, 108, 3838–3841. [Google Scholar] [CrossRef] [Green Version]
- Dey, A.K.; Gel, Y.R.; Poor, H.V. What Network Motifs Tell Us about Resilience and Reliability of Complex Networks. Proc. Natl. Acad. Sci. USA 2019, 116, 19368–19373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milo, R.; Shen-Orr, S.; Itzkovitz, S.; Kashtan, N.; Chklovskii, D.; Alon, U. Network Motifs: Simple Building Blocks of Complex Networks. Science 2002, 298, 824–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menck, P.J.; Heitzig, J.; Kurths, J.; Joachim Schellnhuber, H. How Dead Ends Undermine Power Grid Stability. Nat. Commun. 2014, 5, 3969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, P.; Heitzig, J.; Kurths, J. Detours around Basin Stability in Power Networks. New J. Phys. 2014, 16, 125001. [Google Scholar] [CrossRef]
- Mangan, S.; Alon, U. Structure and Function of the Feed-Forward Loop Network Motif. Proc. Natl. Acad. Sci. USA 2003, 100, 11980–11985. [Google Scholar] [CrossRef] [Green Version]
- Gorochowski, T.E.; Grierson, C.S.; di Bernardo, M. Organization of Feed-Forward Loop Motifs Reveals Architectural Principles in Natural and Engineered Networks. Sci. Adv. 2018, 4, eaap9751. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, E.; de Water, J.V.; Luckhart, S. Malaria-Induced Bacteremia as a Consequence of Multiple Parasite Survival Strategies. Curr. Res. Microb. Sci. 2021, 2, 100036. [Google Scholar] [CrossRef]
- Babatunde, K.A.; Adenuga, O.F. Neutrophils in Malaria: A Double-Edged Sword Role. Front. Immunol. 2022, 13, 922377. [Google Scholar] [CrossRef]
- Dodd, D.; Spitzer, M.H.; Van Treuren, W.; Merrill, B.D.; Hryckowian, A.J.; Higginbottom, S.K.; Le, A.; Cowan, T.M.; Nolan, G.P.; Fischbach, M.A.; et al. A Gut Bacterial Pathway Metabolizes Aromatic Amino Acids into Nine Circulating Metabolites. Nature 2017, 551, 648–652. [Google Scholar] [CrossRef] [Green Version]
- Stough, J.M.A.; Dearth, S.P.; Denny, J.E.; LeCleir, G.R.; Schmidt, N.W.; Campagna, S.R.; Wilhelm, S.W. Functional Characteristics of the Gut Microbiome in C57BL/6 Mice Differentially Susceptible to Plasmodium Yoelii. Front. Microbiol. 2016, 7, 1520. [Google Scholar] [CrossRef] [Green Version]
- Meireles, P.; Brás, D.; Fontinha, D.; Chora, Â.F.; Serre, K.; Mendes, A.M.; Prudêncio, M. Elimination of Hepatic Rodent Plasmodium Parasites by Amino Acid Supplementation. iScience 2020, 23, 101781. [Google Scholar] [CrossRef]
- Loose, L.D.; Silkworth, J.B.; Pittman, K.A.; Benitz, K.F.; Mueller, W. Impaired Host Resistance to Endotoxin and Malaria in Polychlorinated Biphenyl- and Hexachlorobenzene-Treated Mice. Infect. Immun. 1978, 20, 30–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correa, R.; Coronado, L.M.; Garrido, A.C.; Durant-Archibold, A.A.; Spadafora, C. Volatile Organic Compounds Associated with Plasmodium Falciparum Infection in Vitro. Parasites Vectors 2017, 10, 215. [Google Scholar] [CrossRef] [Green Version]
- Kelly, M.; Su, C.-Y.; Schaber, C.; Crowley, J.R.; Hsu, F.-F.; Carlson, J.R.; Odom, A.R. Malaria Parasites Produce Volatile Mosquito Attractants. mBio 2015, 6, e00235-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, B.; Portugal, S.; Tran, T.M.; Gozzelino, R.; Ramos, S.; Gomes, J.; Regalado, A.; Cowan, P.J.; d’Apice, A.J.F.; Chong, A.S.; et al. Gut Microbiota Elicits a Protective Immune Response against Malaria Transmission. Cell 2014, 159, 1277–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Network Features | 8 DPI * | 16 DPI | 24 DPI | 36 DPI | 85 DPI | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Control | Plasmodium-Infected | Control | Plasmodium-Infected | Control | Plasmodium-Infected | Control | Plasmodium-Infected | Control | Plasmodium-Infected | |
| Nodes | 242 (688) ** | 180 (641) | 482 (938) | 571 (1100) | 534 (938) | 473 (1023) | 771 (1059) | 926 (1267) | 463 (926) | 364 (666) |
| Edges | 617 | 399 | 1929 | 2354 | 2673 | 1597 | 4375 | 6299 | 1934 | 1736 |
| Positive | 453 (73.4%) | 245 (61.4%) | 1130 (58.6%) | 1467 (62.3%) | 1597 (59.8%) | 1079 (67.6%) | 2877 (65.8%) | 4169 (66.2%) | 1423 (73.6%) | 1403 (80.8%) |
| Negative | 164 (26.6%) | 154 (38.6%) | 799 (41.4%) | 887 (37.7%) | 1076 (40.2%) | 518 (32.4%) | 1498 (34.2%) | 2130 (33.8%) | 511 (26.4%) | 333 (19.2%) |
| Network diameter | 15 | 16 | 11 | 13 | 9 | 12 | 8 | 13 | 12 | 17 |
| Average degree | 5.099 | 4.433 | 8.004 | 8.245 | 10.011 | 6.753 | 11.349 | 13.605 | 8.354 | 9.538 |
| Weighted degree | 1.928 | 0.827 | 1.124 | 1.67 | 1.651 | 1.946 | 2.897 | 3.649 | 3.254 | 4.84 |
| Average path length | 4.434 | 5.367 | 4.551 | 4.279 | 3.914 | 5.046 | 3.72 | 3.985 | 4.321 | 4.944 |
| Modularity | 1.13 | 2.118 | 2.85 | 2.001 | 2.345 | 1.474 | 1.657 | 1.543 | 1.013 | 0.939 |
| Number of modules | 55 | 49 | 75 | 106 | 59 | 84 | 63 | 61 | 73 | 45 |
| Average clustering coefficient | 0.43 | 0.422 | 0.529 | 0.459 | 0.466 | 0.448 | 0.475 | 0.446 | 0.458 | 0.473 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aželytė, J.; Wu-Chuang, A.; Maitre, A.; Žiegytė, R.; Mateos-Hernández, L.; Obregón, D.; Palinauskas, V.; Cabezas-Cruz, A. Avian Malaria Parasites Modulate Gut Microbiome Assembly in Canaries. Microorganisms 2023, 11, 563. https://doi.org/10.3390/microorganisms11030563
Aželytė J, Wu-Chuang A, Maitre A, Žiegytė R, Mateos-Hernández L, Obregón D, Palinauskas V, Cabezas-Cruz A. Avian Malaria Parasites Modulate Gut Microbiome Assembly in Canaries. Microorganisms. 2023; 11(3):563. https://doi.org/10.3390/microorganisms11030563
Chicago/Turabian StyleAželytė, Justė, Alejandra Wu-Chuang, Apolline Maitre, Rita Žiegytė, Lourdes Mateos-Hernández, Dasiel Obregón, Vaidas Palinauskas, and Alejandro Cabezas-Cruz. 2023. "Avian Malaria Parasites Modulate Gut Microbiome Assembly in Canaries" Microorganisms 11, no. 3: 563. https://doi.org/10.3390/microorganisms11030563