
Effects of Inbreeding on Microbial Community Diversity of Zea mays

Abstract
:1. Introduction
2. Materials and Methods
2.1. Field and Greenhouse Design
2.2. Sample Collection and Processing
2.3. DNA Extraction and Sequencing
2.4. Bioinformatics
2.5. MiniMaize Inoculation Experiment
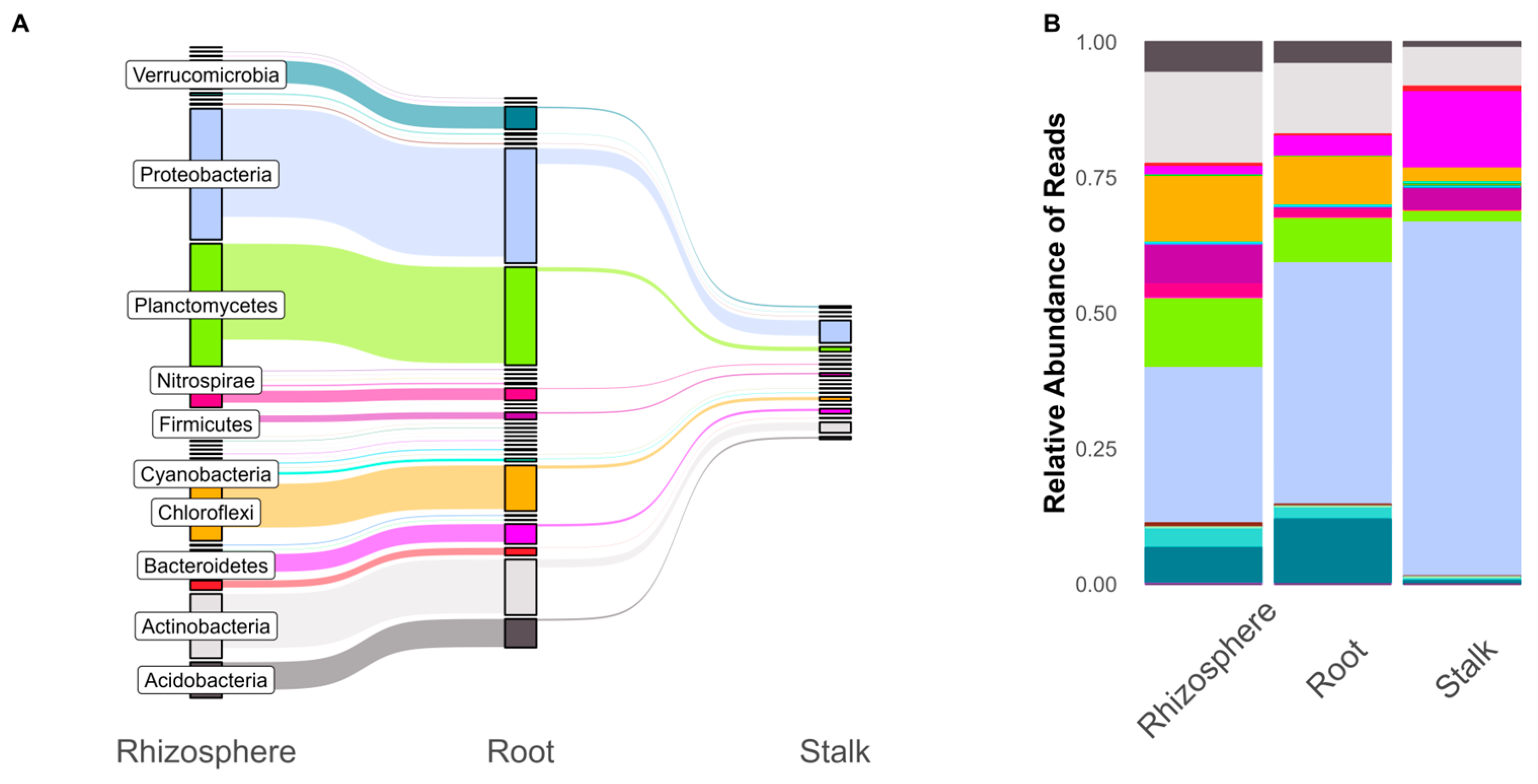
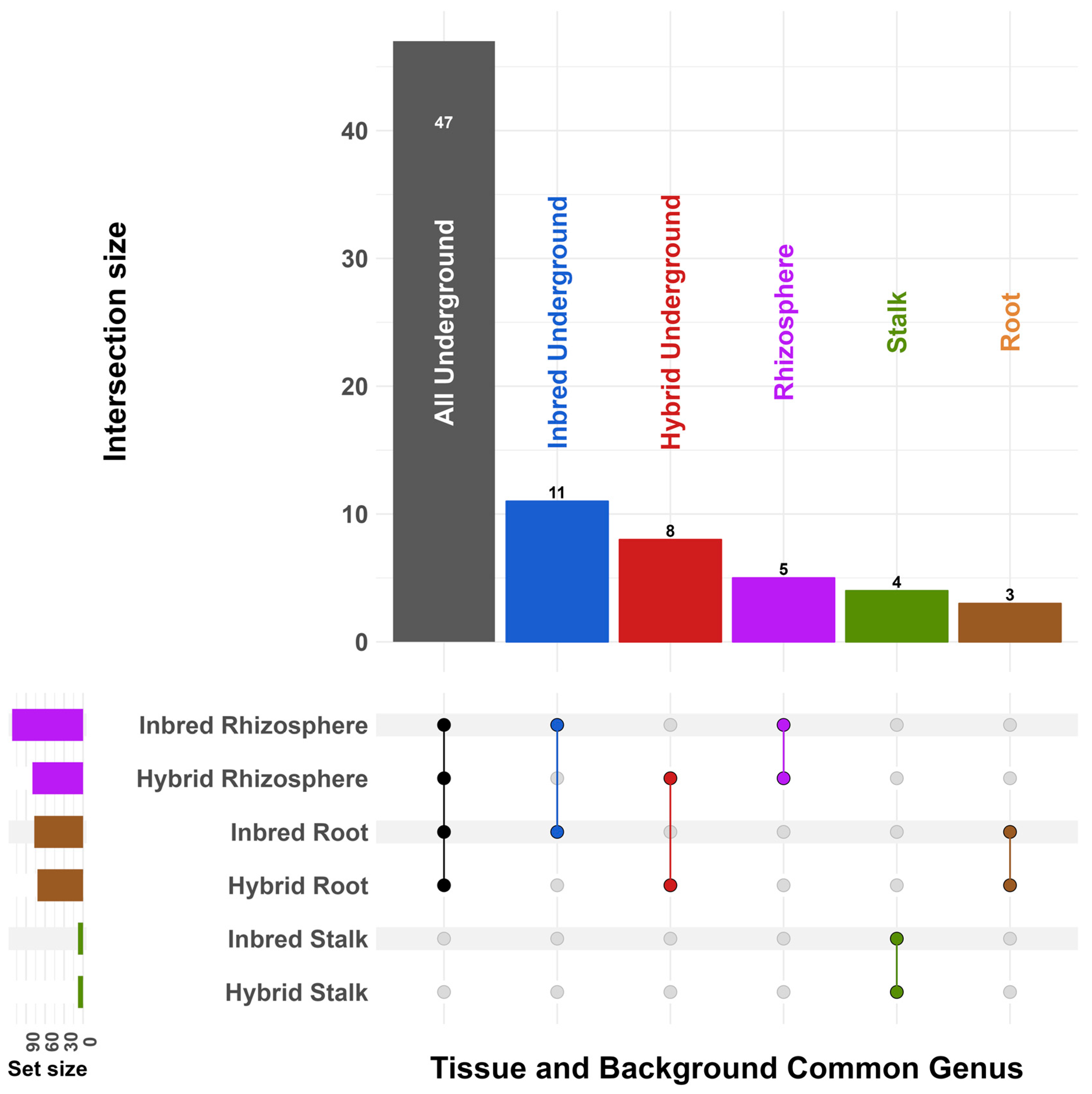
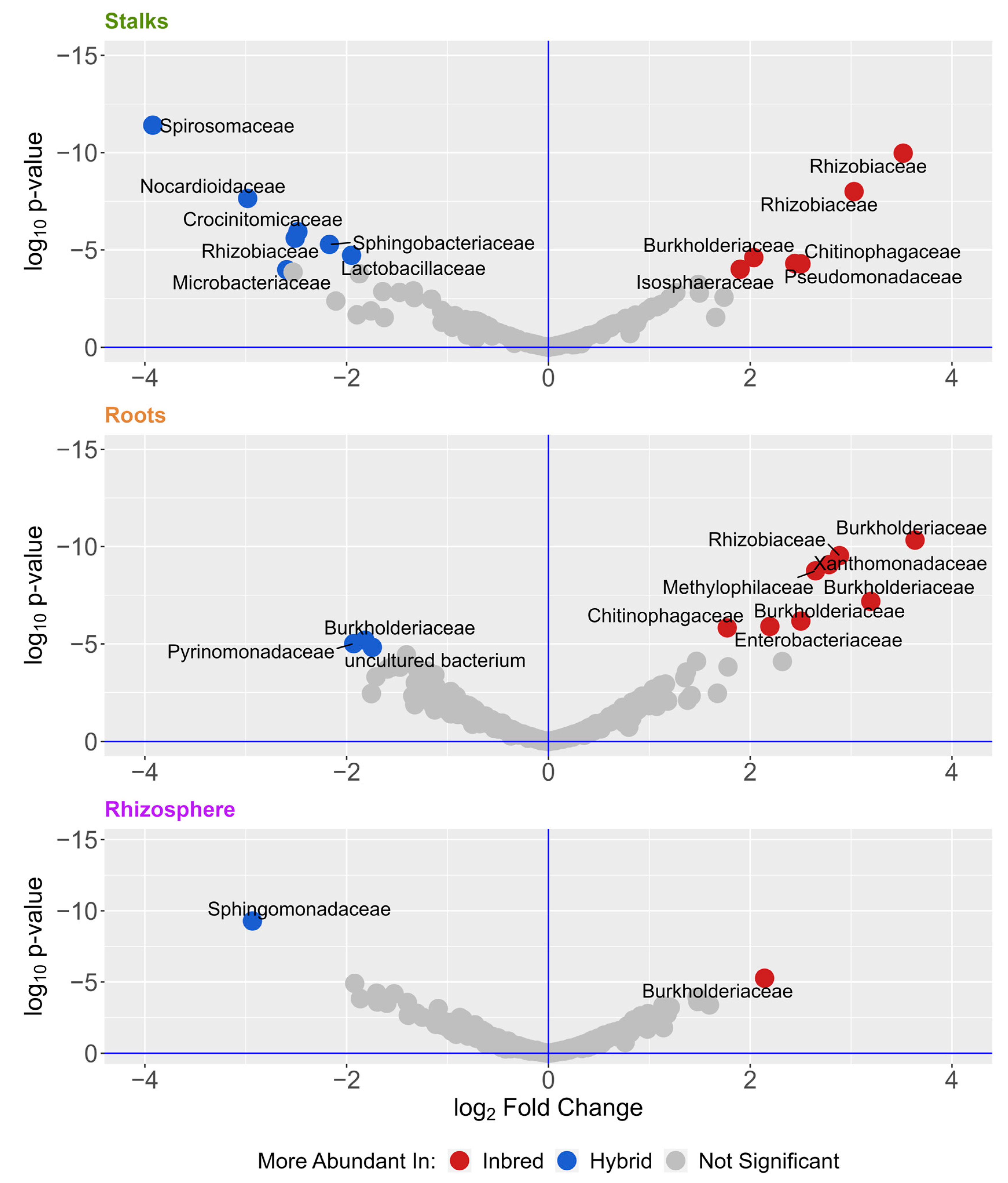
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wallace, J.G.; May, G. Endophytes: The Other Maize Genome. In The Maize Genome; Bennetzen, J., Flint-Garcia, S., Hirsch, C., Tuberosa, R., Eds.; Compendium of Plant Genomes; Springer International Publishing: Cham, Switzerland, 2018; pp. 213–246. ISBN 978-3-319-97427-9. [Google Scholar]
- Compant, S.; Samad, A.; Faist, H.; Sessitsch, A. A review on the plant microbiome: Ecology, functions, and emerging trends in microbial application. J. Adv. Res. 2019, 19, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Peiffer, J.A.; Spor, A.; Koren, O.; Jin, Z.; Tringe, S.G.; Dangl, J.L.; Buckler, E.S.; Ley, R.E. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 6548–6553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innerebner, G.; Knief, C.; Vorholt, J.A. Protection of Arabidopsis thaliana against Leaf-Pathogenic Pseudomonas syringae by Sphingomonas Strains in a Controlled Model System. Appl. Environ. Microbiol. 2011, 77, 3202–3210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cagnano, G.; Lenk, I.; Roulund, N.; Jensen, C.S.; Cox, M.P.; Asp, T. Mycelial biomass and concentration of loline alkaloids driven by complex population structure in Epichloë uncinata and meadow fescue (Schedonorus pratensis). Mycologia 2020, 112, 474–490. [Google Scholar] [CrossRef]
- Rojas, X.; Guo, J.; Leff, J.W.; McNear, D.H.; Fierer, N.; McCulley, R.L. Infection with a Shoot-Specific Fungal Endophyte (Epichloë) Alters Tall Fescue Soil Microbial Communities. Microb. Ecol. 2016, 72, 197–206. [Google Scholar] [CrossRef]
- Passera, A.; Follador, A.; Morandi, S.; Miotti, N.; Ghidoli, M.; Venturini, G.; Quaglino, F.; Brasca, M.; Casati, P.; Pilu, R.; et al. Bacterial Communities in the Embryo of Maize Landraces: Relation with Susceptibility to Fusarium Ear Rot. Microorganisms 2021, 9, 2388. [Google Scholar] [CrossRef]
- Castiglioni, P.; Warner, D.; Bensen, R.J.; Anstrom, D.C.; Harrison, J.; Stoecker, M.; Abad, M.; Kumar, G.; Salvador, S.; D’Ordine, R.; et al. Bacterial RNA Chaperones Confer Abiotic Stress Tolerance in Plants and Improved Grain Yield in Maize under Water-Limited Conditions. Plant Physiol. 2008, 147, 446–455. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, S.S.; Andersen, M.N.; Naveed, M.; Zahir, Z.A.; Liu, F. Interactive effect of biochar and plant growth-promoting bacterial endophytes on ameliorating salinity stress in maize. Funct. Plant Biol. 2015, 42, 770. [Google Scholar] [CrossRef]
- Naveed, M.; Mitter, B.; Reichenauer, T.G.; Wieczorek, K.; Sessitsch, A. Increased drought stress resilience of maize through endophytic colonization by Burkholderia phytofirmans PsJN and Enterobacter sp. FD17. Environ. Exp. Bot. 2014, 97, 30–39. [Google Scholar] [CrossRef]
- Jochum, M.D.; McWilliams, K.L.; Pierson, E.A.; Jo, Y.-K. Host-mediated microbiome engineering (HMME) of drought tolerance in the wheat rhizosphere. PLoS ONE 2019, 14, e0225933. [Google Scholar] [CrossRef] [Green Version]
- Naylor, D.; Degraaf, S.; Purdom, E.; Coleman-Derr, D. Drought and host selection influence bacterial community dynamics in the grass root microbiome. ISME J. 2017, 11, 2691–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldotto, L.E.B.; Olivares, F.L.; Bressan-Smith, R. Structural Interaction Between GFP-Labeled Diazotrophic Endophytic Bacterium Herbaspirillum seropedicae RAM10 and Pineapple Plantlets ‘VitóRia’. Braz. J. Microbiol. 2011, 42, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, E.E.; Secco, V.A.; Moreira, R.S.; dos Santos, O.J.A.P.; Hungria, M.; de Oliveira, A.L.M. Composition and activity of endophytic bacterial communities in field-grown maize plants inoculated with Azospirillum brasilense. Ann. Microbiol. 2015, 65, 2187–2200. [Google Scholar] [CrossRef]
- Young, L.-S.; Hameed, A.; Peng, S.-Y.; Shan, Y.-H.; Wu, S.-P. Endophytic establishment of the soil isolate Burkholderia sp. CC-Al74 enhances growth and P-utilization rate in maize (Zea mays L.). Appl. Soil Ecol. 2013, 66, 40–47. [Google Scholar] [CrossRef]
- Alves, G.C.; Videira, S.S.; Urquiaga, S.; Reis, V.M. Differential plant growth promotion and nitrogen fixation in two genotypes of maize by several Herbaspirillum inoculants. Plant Soil 2014, 387, 307–321. [Google Scholar] [CrossRef]
- Caradonia, F.; Ronga, D.; Catellani, M.; Azevedo, C.V.G.; Terrazas, R.A.; Robertson-Albertyn, S.; Francia, E.; Bulgarelli, D. Nitrogen Fertilisers Shape the Composition and Predicted Functions of the Microbiota of Field-Grown Tomato Plants. bioRxiv 2019, 672162. [Google Scholar]
- Ali, B.; Sabri, A.; Ljung, K.; Hasnain, S. Auxin production by plant associated bacteria: Impact on endogenous IAA content and growth of Triticum aestivum L. Lett. Appl. Microbiol. 2009, 48, 542–547. [Google Scholar] [CrossRef]
- Rivas-Franco, F.; Hampton, J.G.; Narciso, J.; Rostás, M.; Wessman, P.; Saville, D.J.; Jackson, T.A.; Glare, T.R. Effects of a maize root pest and fungal pathogen on entomopathogenic fungal rhizosphere colonization, endophytism and induction of plant hormones. Biol. Control 2020, 150, 104347. [Google Scholar] [CrossRef]
- Kumara, P.M.; Shweta, S.; Vasanthakumari, M.M.; Sachin, N.; Manjunatha, B.L.; Jadhav, S.S.; Ravikanth, G.; Ganeshaiah, K.N.; Shaanker, R.U. Endophytes and Plant Secondary Metabolite Synthesis: Molecular and Evolutionary Perspective. In Advances in Endophytic Research; Springer: Berlin, Germany, 2013; pp. 177–190. [Google Scholar]
- Van Wees, S.C.; Van der Ent, S.; Pieterse, C.M. Plant immune responses triggered by beneficial microbes. Curr. Opin. Plant Biol. 2008, 11, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Oukala, N.; Aissat, K.; Pastor, V. Bacterial Endophytes: The Hidden Actor in Plant Immune Responses against Biotic Stress. Plants 2021, 10, 1012. [Google Scholar] [CrossRef]
- Ma, Y. Beneficial Bacteria for Disease Suppression and Plant Growth Promotion. In Plant-Microbe Interactions in Agro-Ecological Perspectives; Springer: Singapore, 2017; pp. 513–529. ISBN 978-981-10-5812-7. [Google Scholar]
- Zhang, W.; Mason, G.A. Modulating the rhizosphere microbiome by altering the cocktail of root secretions. Plant Physiol. 2022, 188, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Jiang, S.; Jiang, C.; Wu, C.; Gao, M.; Wang, Q. A review of root exudates and rhizosphere microbiome for crop production. Environ. Sci. Pollut. Res. 2021, 28, 54497–54510. [Google Scholar] [CrossRef]
- Wu, L.; Kobayashi, Y.; Wasaki, J.; Koyama, H. Organic acid excretion from roots: A plant mechanism for enhancing phosphorus acquisition, enhancing aluminum tolerance, and recruiting beneficial rhizobacteria. Soil Sci. Plant Nutr. 2018, 64, 697–704. [Google Scholar] [CrossRef]
- Zhalnina, K.; Louie, K.B.; Hao, Z.; Mansoori, N.; da Rocha, U.N.; Shi, S.; Cho, H.; Karaoz, U.; Loqué, D.; Bowen, B.P.; et al. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat. Microbiol. 2018, 3, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Bais, H.P.; Weir, T.L.; Perry, L.G.; Gilroy, S.; Vivanco, J.M. The role of root exudates in rhizosphere interactions with plants and other organisms. Annu. Rev. Plant Biol. 2006, 57, 233–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergelson, J.; Brachi, B.; Roux, F.; Vailleau, F. Assessing the potential to harness the microbiome through plant genetics. Curr. Opin. Biotechnol. 2021, 70, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Dastogeer, K.M.; Tumpa, F.H.; Sultana, A.; Akter, M.A.; Chakraborty, A. Plant microbiome–an account of the factors that shape community composition and diversity. Curr. Plant Biol. 2020, 23, 100161. [Google Scholar] [CrossRef]
- French, E.; Kaplan, I.; Iyer-Pascuzzi, A.; Nakatsu, C.H.; Enders, L. Emerging strategies for precision microbiome management in diverse agroecosystems. Nat. Plants 2021, 7, 256–267. [Google Scholar] [CrossRef]
- Xiong, C.; Zhu, Y.; Wang, J.; Singh, B.; Han, L.; Shen, J.; Li, P.; Wang, G.; Wu, C.; Ge, A.; et al. Host selection shapes crop microbiome assembly and network complexity. New Phytol. 2020, 229, 1091–1104. [Google Scholar] [CrossRef]
- Wallace, J.G.; Kremling, K.A.; Kovar, L.L.; Buckler, E.S. Quantitative Genetics of the Maize Leaf Microbiome. Phytobiomes J. 2018, 2, 208–224. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.R.; Roberts, J.H.; Balint-Kurti, P.; Holland, J.B. Heterosis of leaf and rhizosphere microbiomes in field-grown maize. New Phytol. 2020, 228, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Johnston-Monje, D.; Raizada, M.N. Conservation and Diversity of Seed Associated Endophytes in Zea across Boundaries of Evolution, Ethnography and Ecology. PLoS ONE 2011, 6, e20396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman-reyna, V.; Pinili, D.; Borjaa, F.N.; Quibod, I.; Groen, S.C.; Mulyaningsih, E.S.; Rachmat, A.; Slamet-Loedin, I.H.; Alexandrov, N.; Mauleon, R.; et al. The Rice Leaf Microbiome Has a Conserved Community Structure Controlled by Complex Host-Microbe Interactions; Social Science Research Network: Rochester, NY, USA, 2019. [Google Scholar]
- Kim, H.; Lee, K.K.; Jeon, J.; Harris, W.A.; Lee, Y.-H. Domestication of Oryza species eco-evolutionarily shapes bacterial and fungal communities in rice seed. Microbiome 2020, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Wipf, H.M.L.; Coleman-Derr, D. Evaluating domestication and ploidy effects on the assembly of the wheat bacterial microbiome. PLoS ONE 2021, 16, e0248030. [Google Scholar] [CrossRef] [PubMed]
- Gholizadeh, S.; Mohammadi, S.A.; Salekdeh, G.H. Changes in root microbiome during wheat evolution. BMC Microbiol. 2022, 22, 1–17. [Google Scholar] [CrossRef]
- Veach, A.M.; Morris, R.; Yip, D.Z.; Yang, Z.K.; Engle, N.L.; Cregger, M.A.; Tschaplinski, T.J.; Schadt, C.W. Rhizosphere microbiomes diverge among Populus trichocarpa plant-host genotypes and chemotypes, but it depends on soil origin. Microbiome 2019, 7, 1–15. [Google Scholar] [CrossRef]
- Cordovez, V.; Rotoni, C.; Dini-Andreote, F.; Oyserman, B.; Carrión, V.J.; Raaijmakers, J.M. Successive plant growth amplifies genotype-specific assembly of the tomato rhizosphere microbiome. Sci. Total. Environ. 2021, 772, 144825. [Google Scholar] [CrossRef]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Bennetzen, J.L.; Hake, S. (Eds.) Handbook of Maize; Springer: New York, NY, USA, 2009; ISBN 978-0-387-77862-4. [Google Scholar]
- Corn: USDA ARS. Available online: https://www.ars.usda.gov/oc/timeline/corn/ (accessed on 8 December 2021).
- Corn. Available online: https://www.fas.usda.gov/commodities/corn (accessed on 28 May 2022).
- USDA—National Agricultural Statistics Service—Publications. Available online: https://www.nass.usda.gov/Publications/ (accessed on 28 May 2022).
- Walters, W.A.; Jin, Z.; Youngblut, N.; Wallace, J.G.; Sutter, J.; Zhang, W.; González-Peña, A.; Peiffer, J.; Koren, O.; Shi, Q.; et al. Large-scale replicated field study of maize rhizosphere identifies heritable microbes. Proc. Natl. Acad. Sci. USA 2018, 115, 7368–7373. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.R.; Busby, P.E.; Balint-Kurti, P. Analysis of leaf microbiome composition of near-isogenic maize lines differing in broad-spectrum disease resistance. New Phytol. 2020, 225, 2152–2165. [Google Scholar] [CrossRef]
- Wagner, M.R.; Tang, C.; Salvato, F.; Clouse, K.M.; Bartlett, A.; Vintila, S.; Phillips, L.; Sermons, S.; Hoffmann, M.; Balint-Kurti, P.J.; et al. Microbe-dependent heterosis in maize. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Duvick, D.N. Genetic Contributions to Yield Gains of U.S. Hybrid Maize, 1930 to 1980. In Genetic Contributions to Yield Gains of Five Major Crop Plants; John Wiley & Sons, Ltd.: New York, NY, USA, 1984; pp. 15–47. ISBN 978-0-89118-586-4. [Google Scholar]
- Duvick, D.N. Heterosis: Feeding People and Protecting Natural Resources. In Genetics and Exploitation of Heterosis in Crops; John Wiley & Sons, Ltd.: New York, NY, USA, 1999; pp. 19–29. ISBN 978-0-89118-255-9. [Google Scholar]
- Dewey, L.; Nolan, R. A Guide to Corn Production in Georgia 2018; University of Georgia: Athens, GA, USA, 2018. [Google Scholar]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rrna primers for marine microbiomes with mock communities, time series and Global Field samples. Environ. Microbiol. 2015, 18, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rrna 806r gene primer greatly increases detection of sar11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 8 December 2021).
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 2016, e2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, e00191-16. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Ogle, D.H.; Doll, J.C.; Wheeler, P.; Dinno, A. FSA: Fisheries Stock Analysis. 2022. R Package. Available online: https://CRAN.R-project.org/package=FSA (accessed on 8 December 2021).
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. 2020. R Package. Available online: https://CRAN.R-project.org/package=vegan (accessed on 8 December 2021).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R Package for the Visualization of Intersecting Sets and Their Properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2015, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauvert, C. Psadd: Additions to Phyloseq Package for Microbiome Analysis. 2021. R Package. Available online: https://rdrr.io/github/cpauvert/psadd/ (accessed on 8 December 2021).
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, C.; Brantley, K.; Wallace, J. The Role of Genetic Variation in Maize Response to Beneficial Endophytes. bioRxiv 2021. [Google Scholar] [CrossRef]
- Panke-Buisse, K.; Poole, A.C.; Goodrich, J.K.; E Ley, R.; Kao-Kniffin, J. Selection on soil microbiomes reveals reproducible impacts on plant function. ISME J. 2014, 9, 980–989. [Google Scholar] [CrossRef] [Green Version]
- Mueller, U.G.; Juenger, T.E.; Kardish, M.R.; Carlson, A.L.; Burns, K.; Smith, C.; Marais, D.L.D. Artificial Selection on Microbiomes to Confer Salt-Tolerance to Plants. MSystems 2021, 6, 6. [Google Scholar] [CrossRef]
- Chiu, C.-H.; Jost, L.; Chao, A. Phylogenetic beta diversity, similarity, and differentiation measures based on Hill numbers. Ecol. Monogr. 2014, 84, 21–44. [Google Scholar] [CrossRef]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Lemanceau, P.; Blouin, M.; Muller, D.; Moënne-Loccoz, Y. Let the Core Microbiota Be Functional. Trends Plant Sci. 2017, 22, 583–595. [Google Scholar] [CrossRef]
- Louca, S.; Jacques, S.M.S.; Pires, A.P.F.; Leal, J.S.; Srivastava, D.S.; Parfrey, L.W.; Farjalla, V.F.; Doebeli, M. High taxonomic variability despite stable functional structure across microbial communities. Nat. Ecol. Evol. 2016, 1, 0015. [Google Scholar] [CrossRef] [PubMed]
- McCaw, M.E.; Wallace, J.G.; Albert, P.S.; Buckler, E.S.; Birchler, J.A. Fast-Flowering Mini-Maize: Seed to Seed in 60 Days. Genetics 2016, 204, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston-Monje, D.; Gutiérrez, J.P.; Lopez-Lavalle, L.A.B. Seed-Transmitted Bacteria and Fungi Dominate Juvenile Plant Microbiomes. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Johnston-Monje, D.; Gutiérrez, J.P.; Lopez-Lavalle, L.A.B. Stochastic Inoculum, Biotic Filtering and Species-Specific Seed Transmission Shape the Rare Microbiome of Plants. Life 2022, 12, 1372. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, N.; Guo, X.; Zhang, Y.; Ye, B. Comparative analysis of bacterial community structure in the rhizosphere of maize by high-throughput pyrosequencing. PLoS ONE 2017, 12, e0178425. [Google Scholar] [CrossRef] [Green Version]
- Mehboob, I.; Zahir, Z.; Arshad, M.; Tanveer, A.; Khalid, M. Comparative effectiveness of different rhizobium sp. for improving growth and yield of maize (Zea mays L.). Soil Environ. 2012, 31, 37–46. [Google Scholar]
- Beirinckx, S.; Viaene, T.; Haegeman, A.; Debode, J.; Amery, F.; Vandenabeele, S.; Nelissen, H.; Inzé, D.; Tito, R.; Raes, J.; et al. Tapping into the maize root microbiome to identify bacteria that promote growth under chilling conditions. Microbiome 2020, 8, 54. [Google Scholar] [CrossRef] [PubMed]
- Estrada, G.A.; Baldani, V.L.D.; de Oliveira, D.M.; Urquiaga, S.; Baldani, J.I. Selection of phosphate-solubilizing diazotrophic Herbaspirillum and Burkholderia strains and their effect on rice crop yield and nutrient uptake. Plant Soil 2012, 369, 115–129. [Google Scholar] [CrossRef]
- Tezerji, R.S.; Naveed, M.; Jehl, M.-A.; Sessitsch, A.; Rattei, T.; Mitter, B. The genomes of closely related Pantoea ananatis maize seed endophytes having different effects on the host plant differ in secretion system genes and mobile genetic elements. Front. Microbiol. 2015, 6, 440. [Google Scholar] [CrossRef] [Green Version]
- Doni, F.; Suhaimi, N.S.M.; Irawan, B.; Mohamed, Z.; Mispan, M.S. Associations of Pantoea with Rice Plants: As Friends or Foes? Agriculture 2021, 11, 1278. [Google Scholar] [CrossRef]
- Quecine, M.C.; Araújo, W.L.; Rossetto, P.B.; Ferreira, A.; Tsui, S.; Lacava, P.T.; Mondin, M.; Azevedo, J.L.; Pizzirani-Kleiner, A.A. Sugarcane Growth Promotion by the Endophytic Bacterium Pantoea agglomerans 33.1. Appl. Environ. Microbiol. 2012, 78, 7511–7518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soluch, R.; Hülter, N.F.; Romero Picazo, D.; Özkurt, E.; Stukenbrock, E.H.; Dagan, T. Colonization dynamics of Pantoea agglomerans in the wheat root habitat. Environ. Microbiol. 2021, 23, 2260–2273. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Sun, L.; Huang, Z.; He, L.; Zhang, W.; Chen, Z. Promotion of growth and Cu accumulation of bio-energy crop (Zea mays) by bacteria: Implications for energy plant biomass production and phytoremediation. J. Environ. Manag. 2012, 103, 58–64. [Google Scholar] [CrossRef]
- Piacentino, D.; Grant-Beurmann, S.; Vizioli, C.; Li, X.; Moore, C.F.; Ruiz-Rodado, V.; Lee, M.R.; Joseph, P.V.; Fraser, C.M.; Weerts, E.M.; et al. Gut microbiome and metabolome in a non-human primate model of chronic excessive alcohol drinking. Transl. Psychiatry 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Coles, V.J.; Stukel, M.R.; Brooks, M.T.; Burd, A.; Crump, B.C.; Moran, M.A.; Paul, J.H.; Satinsky, B.M.; Yager, P.L.; Zielinski, B.L.; et al. Ocean biogeochemistry modeled with emergent trait-based genomics. Science 2017, 358, 1149–1154. [Google Scholar] [CrossRef] [Green Version]
- Levy, A.; Salas Gonzalez, I.; Mittelviefhaus, M.; Clingenpeel, S.; Harrera Paredes, S.; Miao, J.; Wang, K.; Devescovi, G.; Stillman, K.; Monteiro, F.; et al. Genomic features of bacterial adaptation to plants. Nat. Genet. 2018, 50, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Bengelsdorf, F.R.; Beck, M.H.; Erz, C.; Hoffmeister, S.; Karl, M.M.; Riegler, P.; Wirth, S.; Poehlein, A.; Weuster-Botz, D.; Dürre, P. Chapter Four—Bacterial Anaerobic Synthesis Gas (Syngas) and CO2+H2 Fermentation. In Advances in Applied Microbiology; Sariaslani, S., Gadd, G.M., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 103, pp. 143–221. [Google Scholar]
- Pang, Z.; Chen, J.; Wang, T.; Gao, C.; Li, Z.; Guo, L.; Xu, J.; Cheng, Y. Linking Plant Secondary Metabolites and Plant Microbiomes: A Review. Front. Plant Sci. 2021, 12. [Google Scholar] [CrossRef]
- Brilli, F.; Loreto, F.; Baccelli, I. Exploiting Plant Volatile Organic Compounds (VOCs) in Agriculture to Improve Sustainable Defense Strategies and Productivity of Crops. Front. Plant Sci. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Scala, A.; Allmann, S.; Mirabella, R.; Haring, M.A.; Schuurink, R.C. Green Leaf Volatiles: A Plant’s Multifunctional Weapon against Herbivores and Pathogens. Int. J. Mol. Sci. 2013, 14, 17781–17811. [Google Scholar] [CrossRef] [Green Version]
- Stolterfoht, H.; Rinnofner, C.; Winkler, M.; Pichler, H. Recombinant Lipoxygenases and Hydroperoxide Lyases for the Synthesis of Green Leaf Volatiles. J. Agric. Food Chem. 2019, 67, 13367–13392. [Google Scholar] [CrossRef]
- Mosquito, S.; Bertani, I.; Licastro, D.; Compant, S.; Myers, M.P.; Hinarejos, E.; Levy, A.; Venturi, V. In Planta Colonization and Role of T6SS in Two Rice Kosakonia Endophytes. Mol. Plant-Microbe Interact. 2020, 33, 349–363. [Google Scholar] [CrossRef]
- Kamat, S.S.; Raushel, F.M. The enzymatic conversion of phosphonates to phosphate by bacteria. Curr. Opin. Chem. Biol. 2013, 17, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Doroghazi, J.R.; Janga, S.C.; Zhang, J.K.; Circello, B.; Griffin, B.M.; Labeda, D.P.; Metcalf, W.W. Diversity and abundance of phosphonate biosynthetic genes in nature. Proc. Natl. Acad. Sci. USA 2013, 110, 20759–20764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, J.A.; Durán, R.V.; De la Rosa, M.A.; Hervás, M. Respiratory cytochrome c oxidase can be efficiently reduced by the photosynthetic redox proteins cytochrome c6 and plastocyanin in cyanobacteria. FEBS Lett. 2005, 579, 3565–3568. [Google Scholar] [CrossRef]
- Deisenhofer, J.; Michel, H. Three-Dimensional Structure of the Reaction Center of Rhodopseudomonas viridis. Photosynth. React. Center 1993, 2, 541–558. [Google Scholar] [CrossRef]
- Cardona, T. A fresh look at the evolution and diversification of photochemical reaction centers. Photosynth. Res. 2014, 126, 111–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperelakis, N. Cell Physiology Source Book; Elsevier Inc.: Amsterdam, The Netherlands, 2012; ISBN 9780123877383. [Google Scholar]
- Kaeppler, S.M.; Parke, J.L.; Mueller, S.M.; Senior, L.; Stuber, C.; Tracy, W.F. Variation among Maize Inbred Lines and Detection of Quantitative Trait Loci for Growth at Low Phosphorus and Responsiveness to Arbuscular Mycorrhizal Fungi. Crop. Sci. 2000, 40, 358–364. [Google Scholar] [CrossRef]
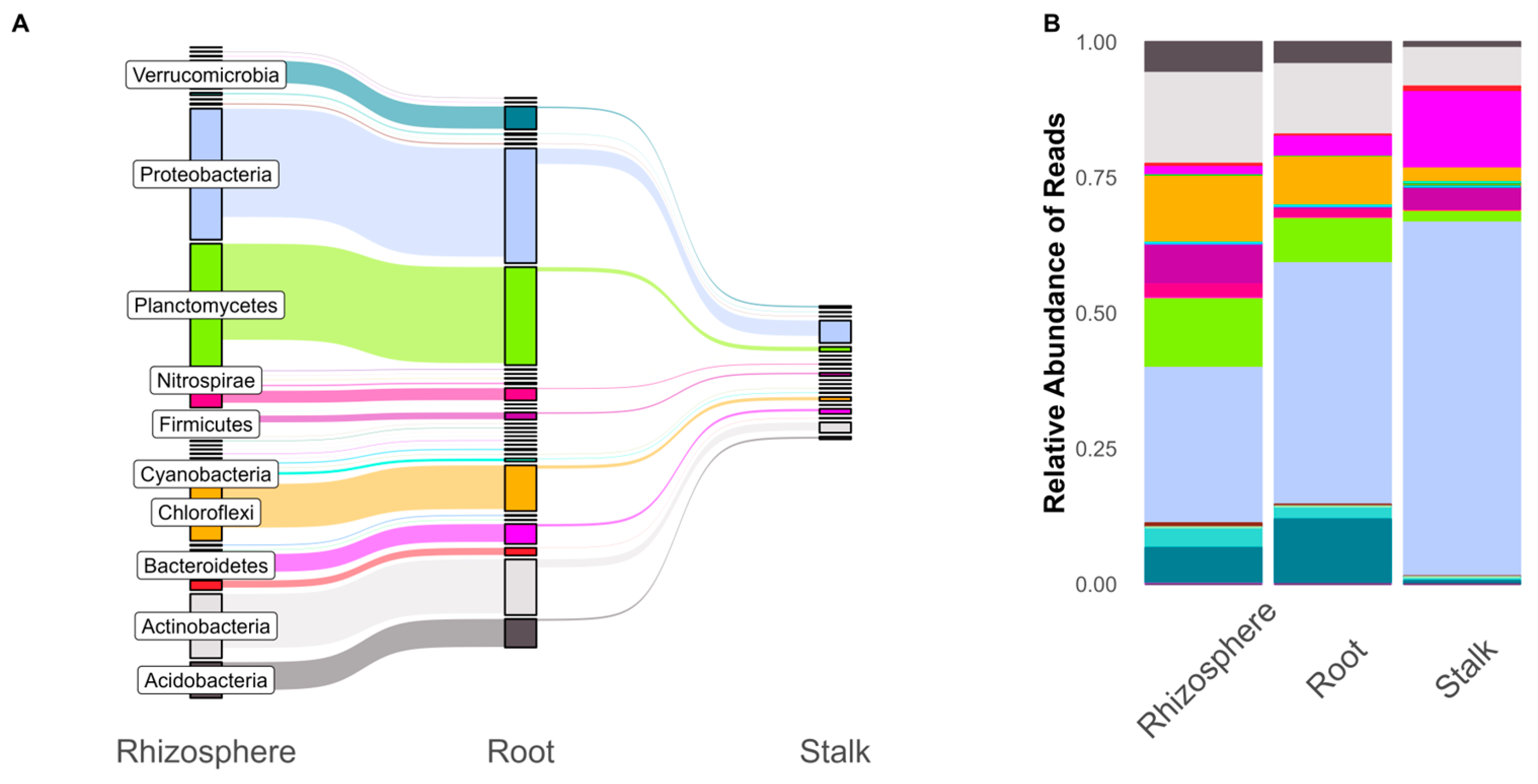
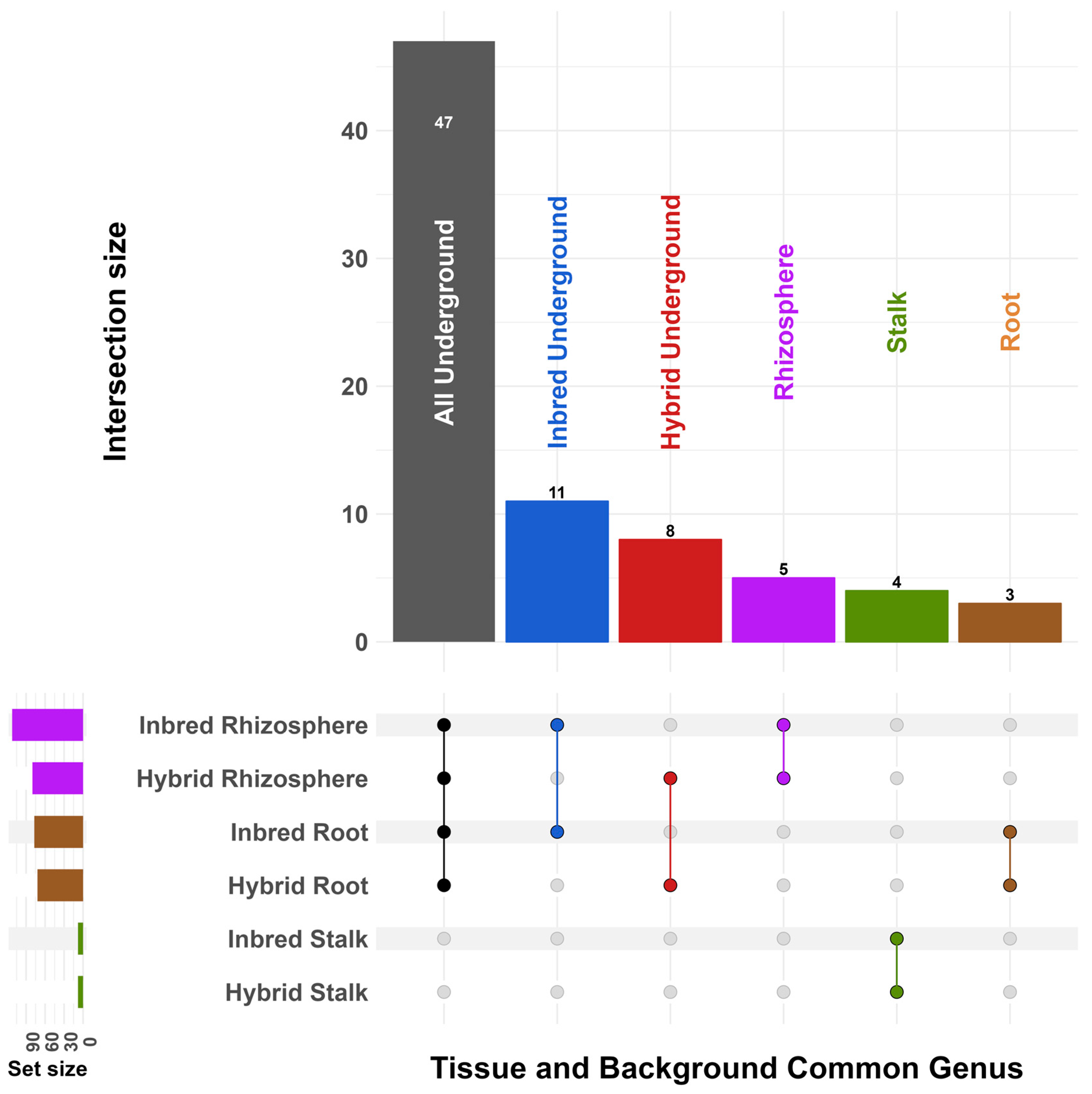
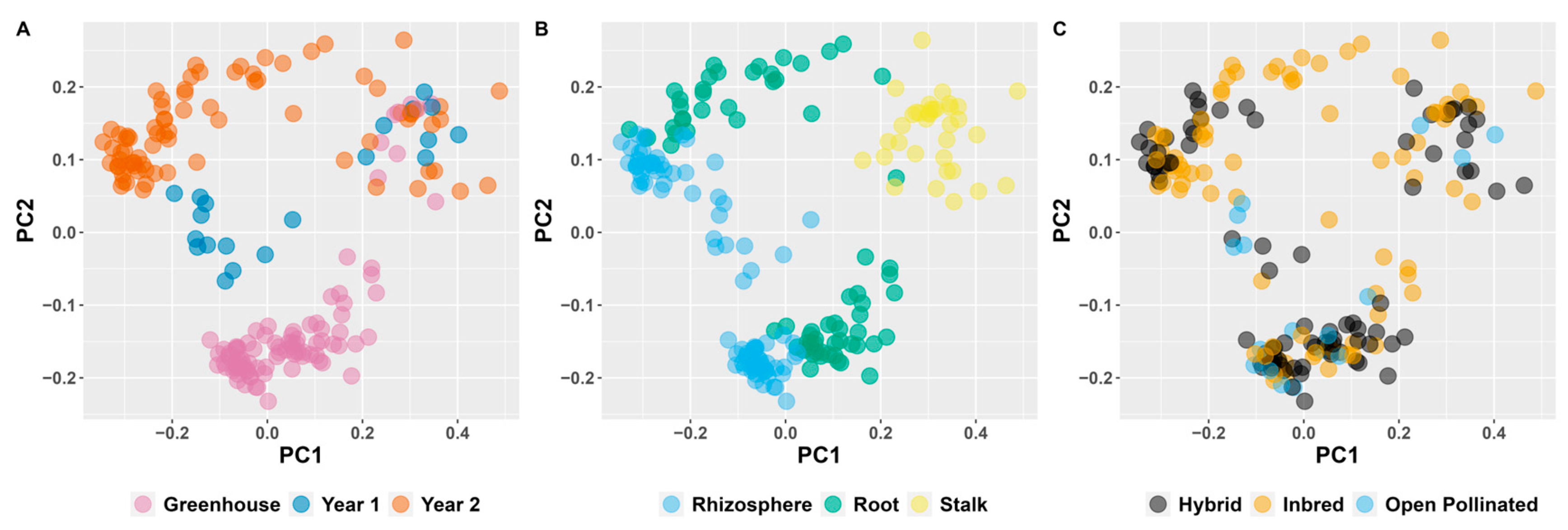
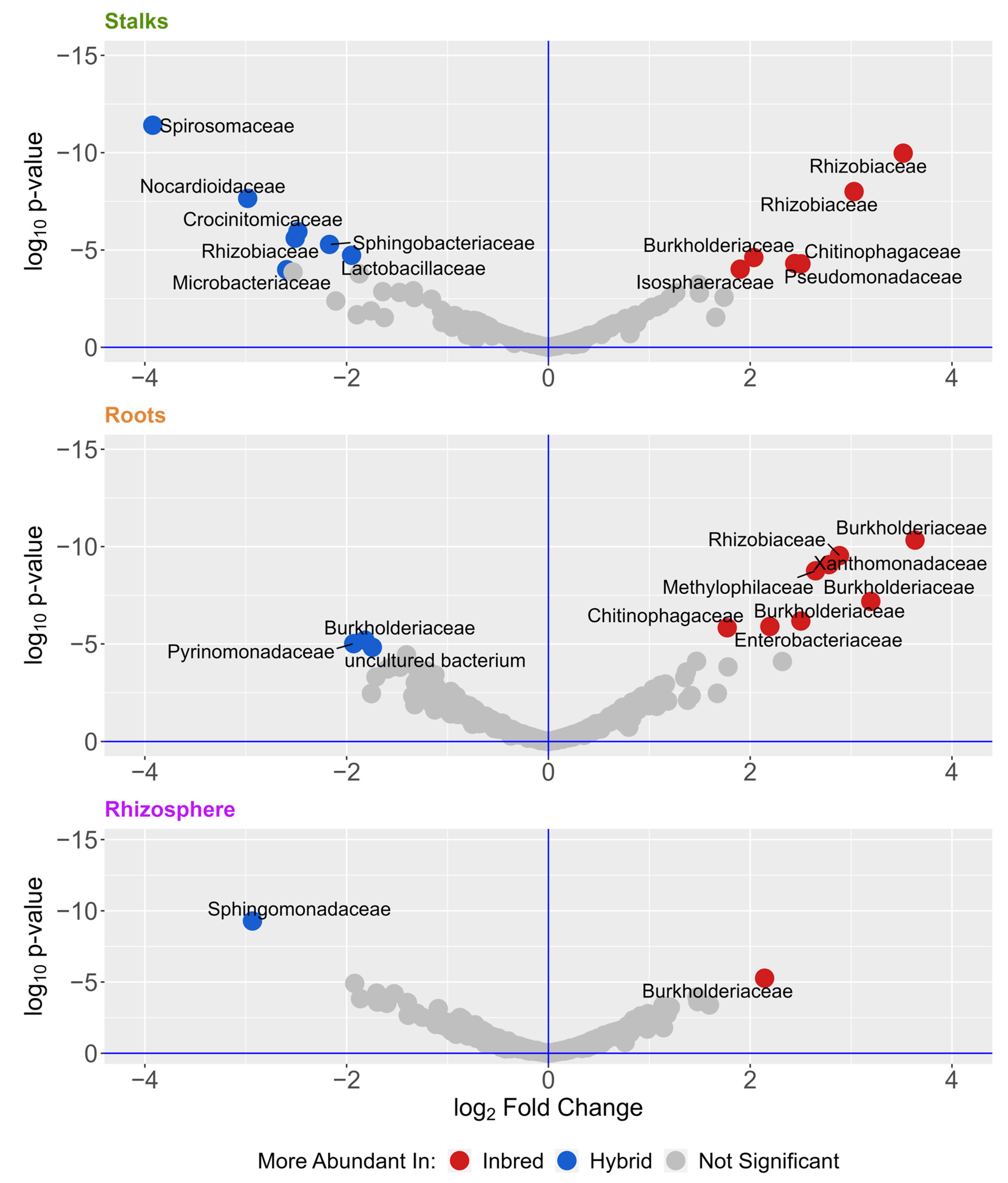

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Maize Genotype | GRIN Accession | Genetic Group | Experiment |
|---|---|---|---|
| CML247 | PI 692141 | Inbred | GH |
| Mo17xPh207 | Hybrid | GH | |
| Mo17 | PI 558532 | Inbred | GH, Year 2 |
| Reid Yellow Dent | PI 222613 | Open Pollinated | GH, Year 1 |
| Ph207 | PI 601005 | Inbred | GH, Year 1, Year 2 |
| Ph207xB73 | Hybrid | GH, Year 1 | |
| B73 | PI 550473 | Inbred | GH, Year 1, Year 2 |
| Oh43 | PI 690332 | Inbred | GH |
| B73xCML247 | Hybrid | GH | |
| B73xOh43 | Hybrid | GH | |
| Mo17xB73 | Hybrid | GH | |
| B73xMo17 | Ames 19097 | Hybrid | GH, Year 2 |
| Hopi_blue | NSL 165817 | Open Pollinated | GH |
| B73xPh207 | Hybrid | GH, Year 1 | |
| DKC70-27 | Hybrid | Year 2 | |
| 903VIP | Hybrid | Year 2 | |
| CML322 | PI 690321 | Inbred | Year 2 |
| HP301 | PI 587131 | Inbred | Year 2 |
| Bloody Butcher | Ames 32345 | Open Pollinated | Year 1 |
| Tissue | Comparison | Number of ASVs |
|---|---|---|
| Compared genetic background | ||
| All | Inbred vs. Hybrid | 61 |
| All | Inbred vs. Open Pollinated | 76 |
| All | Hybrid vs. Open Pollinated | 20 |
| Rhizos | Inbred vs. Hybrid | 2 |
| Rhizos | Inbred vs. Open Pollinated | 8 |
| Rhizos | Hybrid vs. Open Pollinated | 5 |
| Roots | Inbred vs. Hybrid | 11 |
| Roots | Inbred vs. Open Pollinated | 6 |
| Roots | Hybrid vs. Open Pollinated | 2 |
| Stalks | Inbred vs. Hybrid | 14 |
| Stalks | Inbred vs. Open Pollinated | 7 |
| Stalks | Hybrid vs. Open Pollinated | 6 |
| Compared locations | ||
| All | Field vs. Greenhouse | 504 |
| Rhizos | Field vs. Greenhouse | 192 |
| Roots | Field vs. Greenhouse | 182 |
| Stalks | Field vs. Greenhouse | 33 |
| Compared tissues | ||
| - | Stalks vs. Rhizos | 512 |
| - | Stalks vs. Roots | 371 |
| - | Roots vs. Rhizos | 274 |
| Tissue | Comparison | Agglomerated Pathways | Raw Annotations |
|---|---|---|---|
| Compared genetic background | |||
| All | Inbred vs. Hybrid | 1 | 261 |
| All | Inbred vs. OP | 0 | 846 |
| All | Hybrid vs. OP | 0 | 192 |
| Stalks | Inbred vs. Hybrid | 2 | 26 |
| Stalks | Inbred vs. OP | 1 | 334 |
| Stalks | Hybrid vs. OP | 0 | 139 |
| Rhizosphere | Inbred vs. Hybrid | 0 | 28 |
| Rhizosphere | Inbred vs. OP | 0 | 29 |
| Rhizosphere | Hybrid vs. OP | 0 | 67 |
| Root | Inbred vs. Hybrid | 34 | 638 |
| Root | Inbred vs. OP | 0 | 170 |
| Root | Hybrid vs. OP | 0 | 21 |
| Compared locations | |||
| All | Greenhouse vs. Field | 48 | 2994 |
| Stalks | Greenhouse vs. Field | 13 | 724 |
| Root | Greenhouse vs. Field | 108 | 4489 |
| Rhizosphere | Greenhouse vs. Field | 176 | 4966 |
| Compared tissues | |||
| - | Stalks vs. Roots | 139 | 5261 |
| - | Stalks vs. Rhizosphere | 165 | 5901 |
| - | Rhizosphere vs. Root | 71 | 3587 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schultz, C.R.; Johnson, M.; Wallace, J.G. Effects of Inbreeding on Microbial Community Diversity of Zea mays. Microorganisms 2023, 11, 879. https://doi.org/10.3390/microorganisms11040879
Schultz CR, Johnson M, Wallace JG. Effects of Inbreeding on Microbial Community Diversity of Zea mays. Microorganisms. 2023; 11(4):879. https://doi.org/10.3390/microorganisms11040879
Chicago/Turabian StyleSchultz, Corey R., Matthew Johnson, and Jason G. Wallace. 2023. "Effects of Inbreeding on Microbial Community Diversity of Zea mays" Microorganisms 11, no. 4: 879. https://doi.org/10.3390/microorganisms11040879
APA StyleSchultz, C. R., Johnson, M., & Wallace, J. G. (2023). Effects of Inbreeding on Microbial Community Diversity of Zea mays. Microorganisms, 11(4), 879. https://doi.org/10.3390/microorganisms11040879