
Bacterial Microbiota from Lab-Reared and Field-Captured Anopheles darlingi Midgut and Salivary Gland

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection
2.3. Dissection and DNA Extraction
2.4. Library Preparation and Sequencing
2.5. Sequence Processing and Microbial Diversity Analysis
3. Results
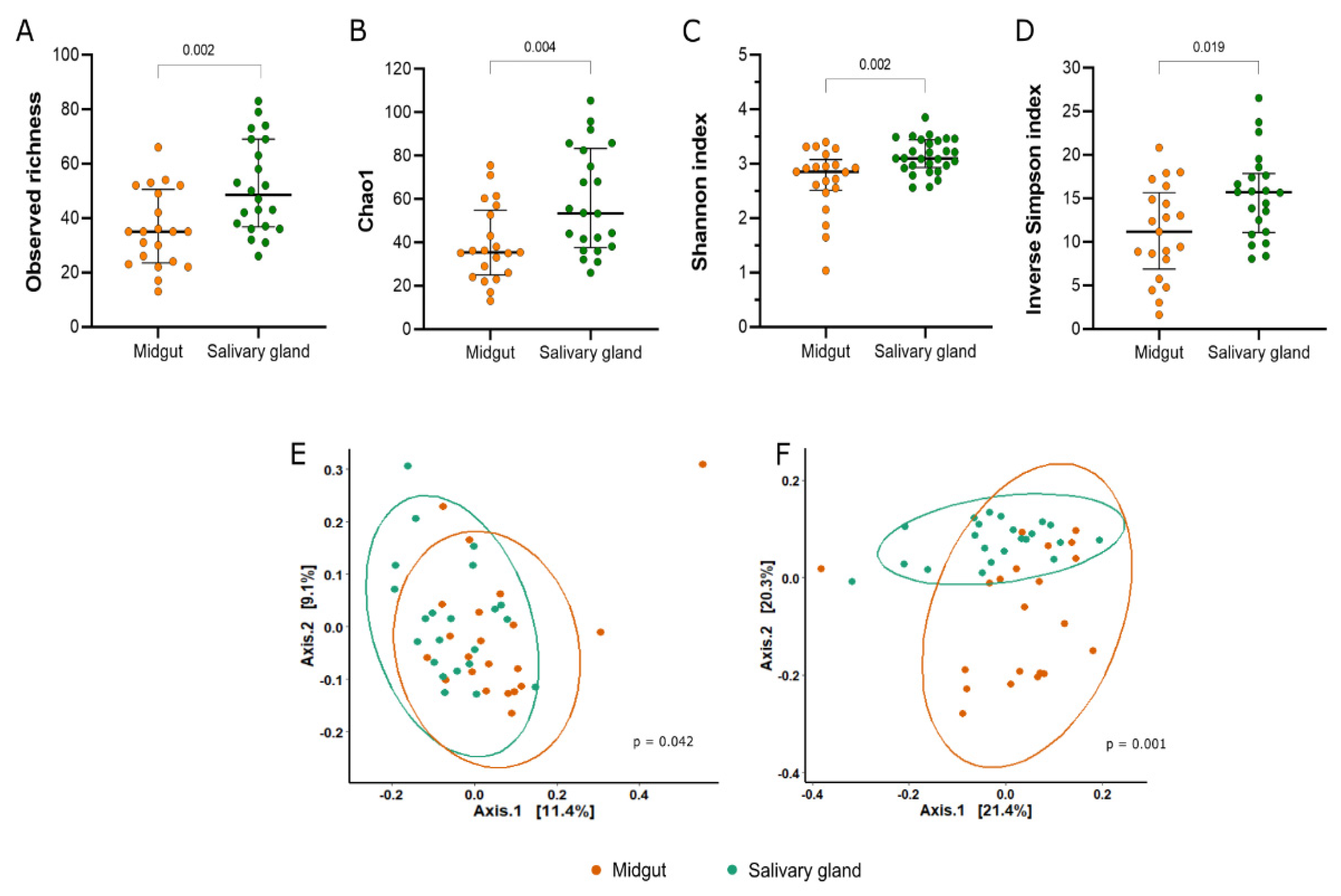
3.1. Diversity of the Bacterial Community from Midgut and Salivary Gland of Lab-Reared Anopheles darlingi
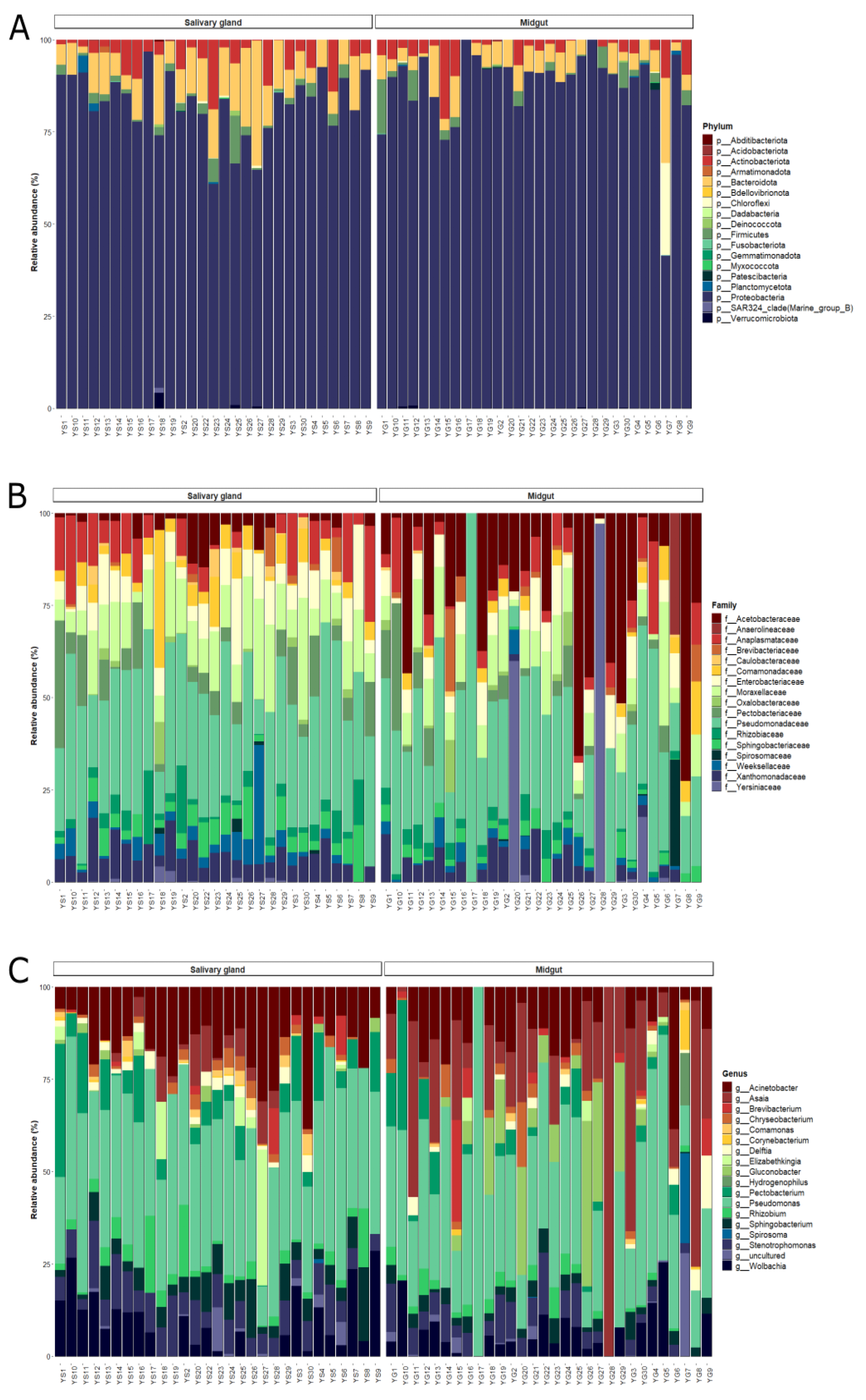
3.2. Composition and Structure of Bacterial Communities from Midgut and Salivary Glands of Lab-Reared Anopheles darlingi
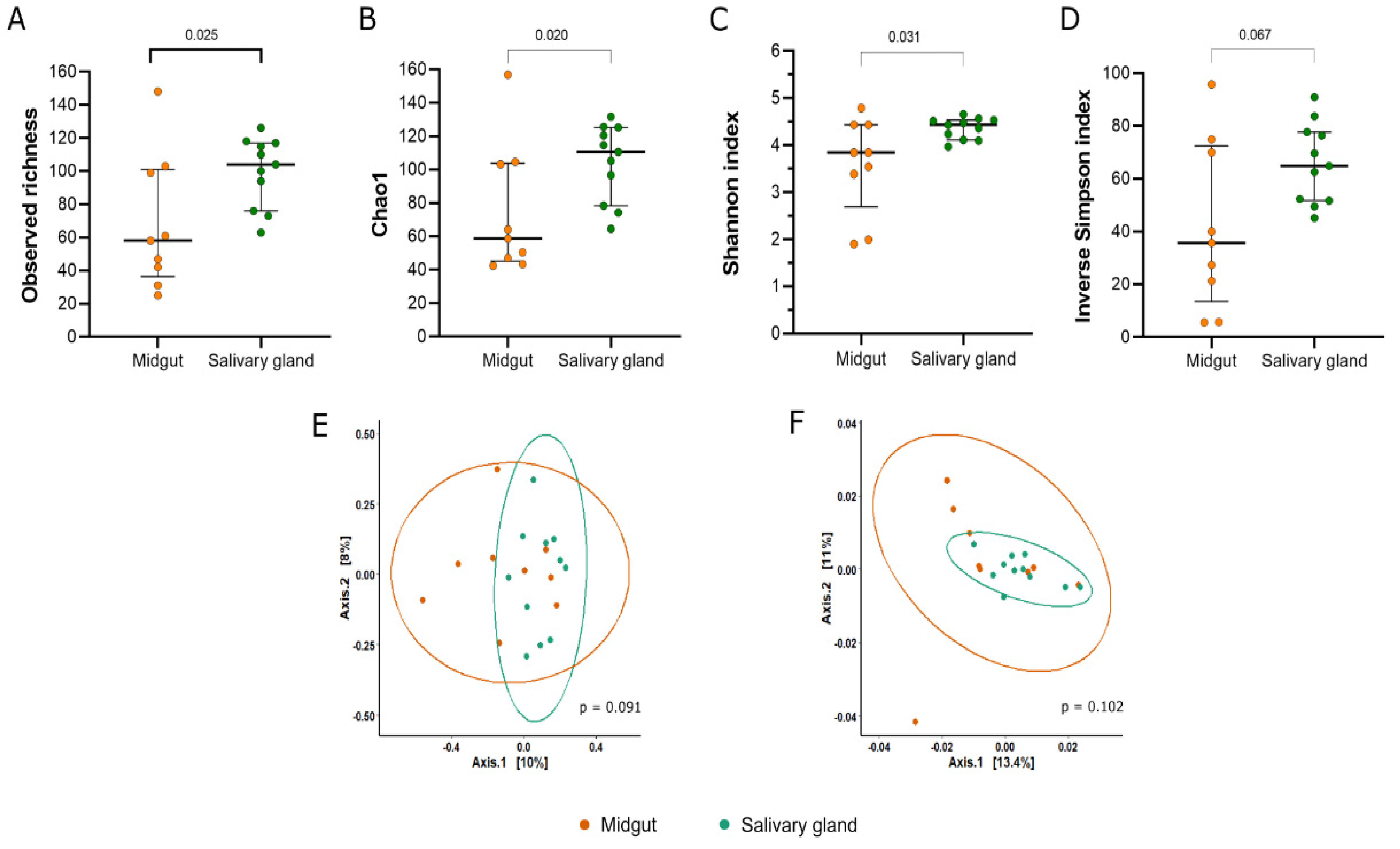
3.3. Diversity of the Bacterial Community from Midgut and Salivary Glands of Field-Captured Anopheles darlingi
3.4. Composition and Structure of Bacterial Communities from Midgut and Salivary Gland of Field-Captured Anopheles darlingi
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vittor, A.Y.; Pan, W.; Gilman, R.H.; Tielsch, J.; Glass, G.; Shields, T.; Glass, G.; Shields, T.; Sánchez-Lozano, W.; Pinedo, V.V.; et al. Linking deforestation to malaria in the Amazon: Characterization of the breeding habitat of the principal malaria vector. Anopheles darlingi. Am. J. Trop. Med. Hyg. 2009, 81, 5–12. [Google Scholar] [PubMed]
- Hiwat, H.; Bretas, G. Ecology of Anopheles darlingi Root with respect to vector importance: A review. Parasit. Vectors. 2011, 4, 177. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, M.P.; Conn, J.E.; Alava, F.; Carrasco-Escobar, G.; Prussing, C.; Bickersmith, S.A.; Sangama, J.L.; Fernandez-Miñope, C.; Guzman, M.; Tong, C.; et al. Higher risk of malaria transmission outdoors than indoors by Nyssorhynchus darlingi in riverine communities in the Peruvian Amazon. Parasites. Vectors. 2019, 12, 374. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.; Saavedra, M.P.; Bickersmith, S.A.; Lainhart, W.; Tong, C.; Alava, F.; Vinetz, J.M.; Coon, J.E. Implications for changes in Anopheles darlingi biting behavior in three communities in the peri-Iquitos region of Amazon Peru. Malar. J. 2015, 14, 290. [Google Scholar] [CrossRef]
- Guégan, M.; Zouache, K.; Démichel, C.; Minard, G.; Van, V.T.; Potier, P.; Mavingui, P.; Moro, C.V. The mosquito holobiont: Fresh insight into mosquito-microbiota interactions. Microbiome. 2018, 6, 49. [Google Scholar] [CrossRef]
- Chandler, J.A.; Liu, R.M.; Bennett, S.N. RNA Shotgun metagenomic sequencing of Northern California (USA) mosquitoes uncovers viruses, bacteria and fungi. Front. Microbiol. 2015, 6, 185. [Google Scholar] [CrossRef]
- Djihinto, O.Y.; Medjigbodo, A.A.; Gangbadja, A.; Saizonou, H.M.; Lagnika, H.O.; Nanmede, D.; Djossou, L.; Bohounton, R.; Sovegnon, P.M.; Fanou, M.J.; et al. Malaria-transmitting vectors microbiota: Overview and interactions with Anopheles mosquito biology. Front. Microbiol. 2022, 13, 891573. [Google Scholar] [CrossRef]
- Vinayagam, S.; Rajendran, D.; Sekar, K.; Renu, K.; Sattu. The microbiota, the malarial parasite, and the mosquito [MMM]—A three-side relationship. Mol. Biochem. Parasitol. 2023, 253, 111543. [Google Scholar] [CrossRef]
- Chouia, B.; Rossi, P.; Epis, S.; Mosca, M.; Ricci, I.; Damiani, C.; Ulissi, U.; Crotti, E.; Daffonchio, D.; Bandi, C.; et al. Delayed larval development in Anopheles mosquitoes deprived of Asaia bacterial symbionts. BMC Microbiol. 2012, 12, S2. [Google Scholar] [CrossRef]
- Coon, K.L.; Vogel, K.J.; Brown, M.R.; Strand, M.R. Mosquitoes rely on their gut microbiota for development. Mol. Ecol. 2014, 23, 2727–2739. [Google Scholar] [CrossRef]
- Wang, X.; Liu, T.; Wu, Y.; Zhong, D.; Zhou, G.; Su, X.; Xu, J.; Sotero, C.F.; Sadruddin, A.A.; Wu, K.; et al. Bacterial microbiota assemblage in Aedes albopictus mosquitoes and its impacts on larval development. Mol. Ecol. 2018, 27, 2972–2985. [Google Scholar] [CrossRef]
- Gaio, A.O.; Gusmão, D.S.; Santos, A.V.; Berbert-Molina, M.A.; Pimenta, P.F.P.; Lemos, F.J.A. Contribution of midgut bacteria to blood digestion and egg production in Aedes aegypti (Diptera: Culicidae) (L.). Parasit. Vectors. 2011, 4, 105. [Google Scholar] [CrossRef]
- Muturi, E.J.; Dunlap, C.; Ramirez, J.L.; Rooney, A.P.; Kim, C.H. Host blood-meal source has a strong impact on gut microbiota of Aedes aegypti. FEMS Microbiol. Ecol. 2019, 95, 213. [Google Scholar] [CrossRef] [PubMed]
- Martinson, V.G.; Strand, M.R. Diet-microbiota interactions alter mosquito development. Front. Microbiol. 2021, 12, 650743. [Google Scholar] [CrossRef]
- Chabanol, E.; Behrends, V.; Prévot, G.; Christophides, G.K.; Gendrin, M. Antibiotic treatment in Anopheles coluzzi affects carbon and nitrogen metabolism. Pathogens 2020, 9, 679. [Google Scholar] [CrossRef]
- Pike, A.; Dong, Y.; Dizaji, N.B.; Gacita, A.; Mongodin, E.F.; Dimopoulos, G. Changes in the microbiota cause genetically modified Anopheles to spread in a population. Science 2017, 357, 1396–1399. [Google Scholar] [CrossRef]
- Dada, N.; Sheth, M.; Liebman, K.; Pinto, J.; Lenhart, A. Whole metagenome sequencing reveals links between mosquito microbiota and insecticide resistance in malaria vectors. Sci. Rep. 2018, 8, 2084. [Google Scholar] [CrossRef] [PubMed]
- Arévalo-Cortés, A.; Mejia-Jaramillo, A.M.; Granada, Y.; Coatsworth, H.; Lowenberger, C.; Triana-Chavez, O. The midgut microbiota of colombian Aedes aegypti populations with different levels of resistance to the insecticide lambda-cyhalothrin. Insects 2020, 11, 584. [Google Scholar] [CrossRef]
- Muturi, E.J.; Dunlap, C.; Smartt, C.T.; Shin, D. Resistance to permethrin alters the gut microbiota of Aedes aegypti. Sci. Rep. 2021, 11, 14406. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Hayatsu, M.; Hosokawa, T.; Fukatsu, T. Symbiont-mediated insecticide resistance. Proc. Natl. Acad. Sci. USA 2012, 109, 8618–8622. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Mandredini, F.; Dimopoulos, G. Implication of the mosquito midgut microbiota in the defense against malaria parasites. PLoS Pathog. 2009, 5, e1000423. [Google Scholar] [CrossRef]
- Cirimotich, C.M.; Dong, Y.; Clayton, A.M.; Sandiford, S.L.; Souza-Neto, J.; Mulenga, M.; Dimopoulos, G. Natural microbe-mediated refractoriness to Plasmodium infection in Anopheles gambiae. Science 2011, 332, 855–858. [Google Scholar] [CrossRef]
- Bascuñán, P.; Niño-Garcia, J.P.; Galeano-Castañeda, Y.; Serre, D.; Correa, M.M. Factors shaping the gut bacterial community assembly in two main Colombian malaria vectors. Microbiome 2018, 6, 148. [Google Scholar] [CrossRef]
- Akorli, J.; Gendrin, M.; Pels, A.P.N.; Yeboah-Manu, D.; Christophides, G.K.; Wilson, M.D. Seasonality and locality affect the diversity of Anopheles gambiae and Anopheles coluzzi midgut microbiota from Ghana. PLoS ONE 2016, 11, e0157529. [Google Scholar] [CrossRef] [PubMed]
- Muturi, E.J.; Ramirez, J.L.; Rooney, A.P.; Kim, C.H. Comparative analysis of gut microbiota of mosquito communities in central Illinois. PLoS Negl. Trop. Dis. 2017, 11, e0005377. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gilbreath, T.M., 3rd; Kukutla, P.; Yan, G.; Xu, J. Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS ONE 2011, 6, e24767. [Google Scholar] [CrossRef] [PubMed]
- Osei-Poku, J.; Mbogo, C.M.; Palmer, W.J.; Jiggins, F.M. Deep sequencing reveals extensive variation in the gut microbiota of wild mosquitoes from Kenya. Mol. Ecol. 2012, 21, 5138–5150. [Google Scholar] [CrossRef]
- Boissière, A.; Tchioffo, M.T.; Bachar, D.; Abate, L.; Marie, A.; Nsango, S.E.; Shahbazkia, H.R.; Awono-Ambene, P.H.; Levashina, E.A.; Christen, R.; et al. Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. PLoS Pathog. 2012, 8, e1002742. [Google Scholar] [CrossRef]
- Sharma, P.; Sharma, S.; Maurya, R.K.; De, T.D.; Thomas, T.; Lata, S.; Singh, N.; Pandey, K.C.; Valecha, N.; Dixit, R. Salivary glands harbor more diverse microbial communities than gut in Anopheles culicifacies. Parasit. Vectors. 2014, 7, 235. [Google Scholar] [CrossRef]
- Tchioffo, M.T.; Boissière, A.; Abate, L.; Nsango, S.E.; Bayibéki, A.N.; Awono-Ambéné, P.; Christen, R.; Gimonneau, G.; Morlais, I. Dynamics of bacterial community composition in the malaria mosquito’s epithelia. Front. Microbiol. 2016, 6, 1500. [Google Scholar] [CrossRef]
- Mancini, M.V.; Damiani, C.; Accoti, A.; Tallarita, M.; Nunzi, E.; Cappelli, A.; Bozic, J.; Catanzani, R.; Rossi, P.; Valzano, M.; et al. Estimating bacteria diversity in different organs of nine species of mosquito by next generation sequencing. BMC Microbiol. 2018, 18, 126. [Google Scholar] [CrossRef]
- Gimonneau, G.; Tchioffo, M.T.; Abate, L.; Boissière, A.; Awono-Ambéné, P.H.; Nsango, S.E.; Christen, R.; Morlais, I. Composition of Anopheles coluzzi and Anopheles gambiae microbiota from larval adult stages. Infect. Genet. Evol. 2014, 28, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, T.M.P.; Sanabani, S.S.; Sallum, M.A.M. Bacterial diversity associated with the abdomens of naturally Plasmodium-infected and non-infected Nyssorhynchus darlingi. BMC Microbiol. 2020, 20, 180. [Google Scholar] [CrossRef]
- Oliveira, T.M.P.; Sanabani, S.S.; Sallum, M.A.M. Asaia (Rhodospirillales: Acetobacteraceae) and Serratia (Enterobacterales: Yersiniaceae) associated with Nyssorhynchus braziliensis and Nyssorhynchus darlingi (Diptera: Culicidae). Rev. Bras. Entomol. 2020, 64, e20190010. [Google Scholar] [CrossRef]
- Rocha, E.M.; Marinotti, O.; Serrão, D.M.; Correa, L.V.; Katak, R.M.; Oliveira, J.C.; Muniz, V.A.; Oliveira, M.R.; Neto, J.F.N.; Pessoa, M.C.F.; et al. Cultured bacteria associated with Anopheles darlingi and their paratransgenesis potential. Malaria. J. 2021, 20, 40. [Google Scholar] [CrossRef]
- Arruda, A.; Ferreira, G.E.M.; Júnior, A.S.; Matos, N.B.; Carvalho, T.S.; Ozaki, L.S.; Stabeli, R.G.; Silva, A.A.E. Diversity of culturable bacteria isolated from the feces of wild Anopheles darlingi (Diptera: Culicidae) mosquitoes from the Brazilian Amazon. J. Med. Entomol. 2021, 58, 1900–1907. [Google Scholar] [CrossRef]
- Tenerius, O.; Oliveira, C.D.; Pinheiro, W.D.; Tadei, W.P.; James, A.A.; Marinotti, O. 16S rRNA gene sequences from bacteria associated with adult Anopheles darlingi (Diptera: Culicidae) mosquitoes. J. Med. Entomol. 2008, 45, 172–175. [Google Scholar] [CrossRef]
- Araujo, M.S.; Andrade, A.O.; Santos, N.A.C.; Pereira, D.B.; Costa, G.S.; Paulo, P.F.M.; Rios, C.T.; Moreno, M.; Pereira-da-Silva, L.H.; Medeiros, J.F. Brazil’s first free-mating laboratory colony of Nyssorhynchus darlingi. Rev. Soc. Med. Trop. 2019, 52, e20190159. [Google Scholar] [CrossRef]
- Consoli, R.A.; Oliveira, R.L. Principais Mosquitos de Importância Sanitária no Brasil; FIOCRUZ: Rio de Janeiro, Brazil, 1994; p. 228. [Google Scholar]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Pro. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribossomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Robeson II, M.S.; O’Rourke, D.R.; Kaehler, B.D.; Ziemski, M.; Dillon, M.R.; Foster, J.T.; Bokulich, N.A. RESCRIPt: Reproducible sequence taxonomy reference database management for the masses. PLoS Comput. Biol. 2021, 17, e1009581. [Google Scholar] [CrossRef]
- Berhanu, A.; Abera, A.; Nega, D.; Mekasha, S.; Fentaw, S.; Assefa, A.; Fentaw, S.; Assefa, A.; Gebrewolde, G.; Wuletaw, Y.; et al. Isolation and identification of microflora from the midgut and salivary glands of Anopheles species in malaria endemic areas of Ethiopia. BMC Microbiol. 2019, 19, 85. [Google Scholar] [CrossRef]
- Finney, C.A.; Kamhawi, S.; Wasmuth, J.D. Does the arthropod microbiota impact the establishment of vector-borne diseases in mammalian hosts? PLoS Pathog. 2015, 11, e1004646. [Google Scholar] [CrossRef]
- Ngo, C.T.; Romano-Bertrand, S.; Manguin, S.; Jumas-Bilak, E. Diversity of the bacterial microbiota of Anopheles mosquitoes from Binh Phuoc Province, Vietnam. Front. Microbiol. 2016, 7, 2095. [Google Scholar] [CrossRef]
- Barnard, K.; Jeanrenaud, A.C.S.N.; Brooke, B.D.; Oliver, S.V. The contribution of gut bacteria to insecticide resistance and the life histories of the major malaria vector Anopheles arabiensis (Diptera: Culicidae). Sci. Rep. 2019, 9, 9117. [Google Scholar] [CrossRef] [PubMed]
- Bahia, A.C.; Dong, Y.; Blumberg, B.J.; Mlambo, G.; Tripathi, A.; Benmarzouk-Hidalgo, O.J.; Chandra, R.; Dimopoulos, G. Exploring Anopheles gut bacteria for Plasmodium blocking activity. Environ. Microbiol. 2014, 16, 2980–2994. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.M.; Maier, W.A.; Rottok, M.; Becker-Feldmann. Concomitant infections of Anopheles stephensi with Plasmodium berghei and Serratia marcescens: Additive detrimental effects. Zentralbl. Backteriol. Mikrobiol. Hyg. A 1987, 266, 155–166. [Google Scholar] [CrossRef]
- Gonzalez-Ceron, L.; Santillan, F.; Rodriguez, M.H.; Mendez, D.; Hernandez-Avila, J.E. Bacteria in midguts of field-collected Anopheles albimanus block Plasmodium vivax sporogonic development. J. Med. Entomol. 2003, 40, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Barletta, A.B.F.; Trisnadi, N.; Ramirez, J.L.; Barillas-Mury, C. Mosquito midgut prostaglandin release establishes systemic immune priming. iScience 2019, 19, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Song, X.; Wang, J. Gut microbiota is essential in PGRP-LA regulated immune protection against Plasmodium berghei infection. Parasit. Vectors. 2020, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Gendrin, M.; Christophides, G.K. The peptidoglycan recognition proteins PGRLA and PGRPLB regulate Anopheles immunity to bacteria and affect infection by Plasmodium. J. Innate. Immun. 2017, 9, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, F.H.; Gendrin, M.; Wyer, C.A.S.; Christophides, G.K. Microbiota-induced peritrophic matrix regulates midgut homeostasis and prevents systemic infection of malaria vector mosquitoes. PLoS Pathog. 2017, 13, e1006391. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wang, M.; Dong, L.; Zhu, H.; Wang, J. PGRP-LD mediates A. stephensi vector competency by regulating homeostasis of microbiota-induced peritrophic matrix synthesis. PLoS Pathog. 2018, 14, e1006899. [Google Scholar] [CrossRef]
- Manguin, S.; Ngo, C.T.; Tainchum, K.; Juntarajumnong, W.; Chareonviriyaphap, T.; Michon, A.L.; Bilak-Jumas, E. Bacterial biodiversity in midgut of Anopheles mosquitoes, malaria vector in Southeast Asia. In Anopheles Mosquitoes—New Insights into Malaria Vectors; Manguin, S., Ed.; Intech Open: London, UK, 2013; pp. 549–576. [Google Scholar] [CrossRef]
- Zoure, A.A.; Sare, A.R.; Yameogo, F.; Somda, Z.; Massart, S.; Badolo, A.; Francis, F. Bacterial communities associated with the midgut microbiota of wild Anopheles gambiae complex in Burkina Faso. Mol. Biol. Rep. 2020, 47, 211–224. [Google Scholar] [CrossRef]
- Galeano-Castañeda, Y.; Bascuñán, P.; Serre, D.; Correa, M.M. Trans-stadial fate of the gut bacterial microbiota in Anopheles albimanus. Acta. Trop. 2020, 201, 105204. [Google Scholar] [CrossRef]
- Alonso, D.P.; Mancini, M.V.; Damiani, C.; Cappelli, A.; Ricci, I.; Alvarez, M.V.N.; Bandi, C.; Ribolla, P.E.M.; Favia, G. Genome reduction in the mosquito symbiont Asaia. Genome. Biol. Evol. 2019, 11, 1–10. [Google Scholar] [CrossRef]
- Favia, G.; Ricci, I.; Damiani, C.; Raddadi, N.; Crotti, E.; Marzorati, M.; Rizzi, A.; Urso, R.; Brusetti, L.; Borin, S.; et al. Bacteria of the genus Asaia stably associate with Anopheles stephensi, an Asian malarial mosquito vector. Proc. Natl. Acad. Sci. USA 2007, 104, 9047–9051. [Google Scholar] [CrossRef]
- Favia, G.; Ricci, I.; Marzorati, M.; Negri, I.; Alma, A.; Sacchi, L.; Bandi, C.; Daffonchio, D. Bacteria of the genus Asaia: A potential paratransgenic weapon against malaria. In Transgenesis and the Management of Vector-Borne Disease; Aksoy, S., Ed.; Springer: New York, NY, USA, 2008; Volume 627, pp. 49–59. [Google Scholar] [CrossRef]
- Crotti, E.; Damiani, C.; Pajoro, M.; Gonella, E.; Rizzi, A.; Ricci, I.; Negri, I.; Scuppa, P.; Rossi, P.; Ballarini, P.; et al. Asaia, a versatile acetic acid bacterial symbiont, capable of cross-colonizing insects of phylogenetically distant genera and orders. Environ. Microbiol. 2009, 11, 3252–3264. [Google Scholar] [CrossRef]
- Damiani, C.; Ricci, I.; Crotti, E.; Rossi, P.; Rizzi, A.; Scuppa, P.; Capone, A.; Ulissi, U.; Epis, S.; Genchi, M.; et al. Mosquito-bacteria symbiosis: The case of Anopheles gambiae and Asaia. Micro. Ecol. 2010, 60, 644–654. [Google Scholar] [CrossRef]
- Gonella, E.; Crotti, E.; Rizzi, A.; Mandrioli, M.; Favia, G.; Daffonchio, D.; Alma, A. Horizontal transmission of the symbiotic bacterium Asaia sp. In the leafhopper Scaphoideus titanus ball (Hemiptera: Cicadellidae). BMC Microbiol. 2012, 12, S4. [Google Scholar] [CrossRef] [PubMed]
- Bongio, N.J.; Lampe, D.J. Inhibition of Plasmodium berghei development in mosquitoes by effector proteins secreted from Asaia sp. Bacteria using a novel native secretion signal. PLoS ONE 2015, 10, e0143541. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, A.; Damiani, C.; Mancini, M.V.; Valzano, M.; Rossi, P.; Serrao, A.; Ricci, I.; Favia, G. Asaia activate imune genes in mosquito eliciting an anti-Plasmodium response: Implications in malaria control. Front. Genet. 2019, 10, 836. [Google Scholar] [CrossRef] [PubMed]
- Ayala, D.; Akone-Ella, O.; Rahola, N.; Kengne, P.; Ngangue, M.F.; Mezeme, F.; Makanga, B.K.; Nigg, M.; Costantini, C.; Simard, F.; et al. Natural Wolbachia infections are common in the major malaria vectors in Central Africa. Evol. Appl. 2019, 12, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Chrostek, E.; Gerth, M. Is Anopheles gambiae a natural host of Wolbachia? mBio 2019, 10, e00784-19. [Google Scholar] [CrossRef] [PubMed]
- Prussing, C.; Saavedra, M.P.; Bickersmith, S.A.; Alava, F.; Guzmán, M.; Manrique, E.; Carrasco-Escobar, G.; Moreno, M.; Gamboa, D.; Vinetz, J.M.; et al. Malaria vector species in Amazonian Peru co-occur in larval habitats but have distinct larval microbial communities. PLoS Negl. Trop. Dis. 2019, 13, e0007412. [Google Scholar] [CrossRef]
- Chen, S.; Bagdasarian, M.; Walker, E.D. Elizabethkingia anophelis: Molecular manipulation and interactions with mosquito hosts. Appl. Environ. Microbiol. 2015, 81, 2233–2243. [Google Scholar] [CrossRef]
- Bisi, D.C.; Lampe, D.J. Secretion of anti-Plasmodium effector proteins from a natural Pantoea agglomerans isolate by using PelB and HlyA secretion signals. Appl. Environ. Microbiol. 2011, 77, 4669–4675. [Google Scholar] [CrossRef]
- Bando, H.; Okado, K.; Guelbeogo, W.M.; Badolo, A.; Aonuma, H.; Nelson, B.; Fukumoto, S.; Xuan, X.; Sagnon, N.F.; Kanuka, H. Intra-specific diversity of Serratia marcescens in Anopheles mosquito midgut defines Plasmodium transmission capacity. Sci. Rep. 2013, 3, 1641. [Google Scholar] [CrossRef]
- Bai, L.; Wang, L.; Vega-Rodríguez, J.; Wang, G.; Wang, S. A gut symbiotic bacterium Serratia marcescens renders mosquito resistance to Plasmodium infection through activation of mosquito immune responses. Front. Microbiol. 2019, 10, 1580. [Google Scholar] [CrossRef]
- Nilsson, L.K.J.; Oliveira, M.R.; Marinotti, O.; Rocha, E.M.; Hakansson, S.; Tadei, W.; Souza, A.Q.L.; Terenius, O. Characterization of bacterial communities in breeding waters of Anopheles darlingi in Manaus in the Amazon basin malaria—Endemic area. Microb. Ecol. 2019, 78, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.; Tong, C.; Guzmán, M.; Chuquiyauri, R.; Llanos-Cuentas, A.; Rodriguez, H.; Gamboa, D.; Meister, S.; Winzeler, E.A.; Maguina, P.; et al. Infection of laboratory-colonized Anopheles darlingi mosquitoes by Plasmodium vivax. Am. J. Trop. Med. Hyg. 2014, 90, 612–616. [Google Scholar] [CrossRef]
- Santos, N.A.C.; Magi, F.N.; Andrade, A.O.; Bastos, A.S.; Pereira, S.S.; Medeiros, J.F.; Araujo, M.S. Assessment of antibiotic treatment on Anopheles darlingi survival and susceptibility to Plasmodium vivax. Front. Microbiol. 2022, 13, 971083. [Google Scholar] [CrossRef] [PubMed]
- Bahia, A.C.; Oliveira, J.H.; Kubota, M.S.; Araújo, H.R.; Lima, J.B.; Ríos-Velásquez, C.M.; Lacerda, M.V.G.; Oliveira, P.L.; Traub-Csekö, Y.M.; Pimenta, P.F. The role of reactive oxygen species in Anopheles aquasalis response to Plasmodium vivax infection. PLoS ONE 2013, 8, e57014. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Rani, J.; Chauhan, C.; Kumari, S.; Tevatiya, S.; De, T.D.; Savargaonkar, D.; Pandey, K.C.; Dixit, R. Altered gut microbiota and immunity defines Plasmodium vivax survival in Anopheles stephensi. Front. Immunol. 2020, 11, 609. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.M.; Katak, R.M.; Oliveira, J.C.; Araujo, M.S.; Carlos, B.C.; Galizi, R.; Tripet, F.; Marinotti, O.; Souza-Neto, J.A. Vector-focused approaches to curb malaria transmission in the Brazilian Amazon: An overview of current and future challenges and strategies. Trop. Med. Infect. Dis. 2020, 5, 161. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prevalence (%) | ||
|---|---|---|
| Genus | Salivary Gland | Midgut |
| Acinetobacter | 100 | 86.6 |
| Pseudomonas | 100 | 96.6 |
| Stenotrophomonas | 96.5 | 73.3 |
| Rhizobium | 93.1 | 66.6 |
| Sphingobacterium | 89.6 | 83.3 |
| Wolbachia | 79.3 | 70 |
| Chryseobacterium | 72.4 | 66.6 |
| Pectobacterium | 72.4 | 56.6 |
| Delftia | 68.9 | - |
| Staphylococcus | 55.1 | - |
| Asaia | - | 80 |
| Gluconobacter | - | 53.3 |
| Prevalence (%) | ||
|---|---|---|
| Genus | Salivary Gland | Midgut |
| Streptococcus | 78.9 | 55.5 |
| Corynebacterium | 57.8 | 61.1 |
| Acinetobacter | 63.1 | 55.5 |
| Pseudomonas | 68.4 | - |
| Staphylococcus | 63.1 | - |
| Rubrobacter | 63.1 | - |
| Bacillus | 57.8 | - |
| Massilia | 52.6 | - |
| Delftia | 52.6 | - |
| Escherichia-Shigella | 52.6 | - |
| Cutibacterium | 52.6 | - |
| Atopococcus | 52.6 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, N.A.C.d.; Carvalho, V.R.d.; Souza-Neto, J.A.; Alonso, D.P.; Ribolla, P.E.M.; Medeiros, J.F.; Araujo, M.d.S. Bacterial Microbiota from Lab-Reared and Field-Captured Anopheles darlingi Midgut and Salivary Gland. Microorganisms 2023, 11, 1145. https://doi.org/10.3390/microorganisms11051145
Santos NACd, Carvalho VRd, Souza-Neto JA, Alonso DP, Ribolla PEM, Medeiros JF, Araujo MdS. Bacterial Microbiota from Lab-Reared and Field-Captured Anopheles darlingi Midgut and Salivary Gland. Microorganisms. 2023; 11(5):1145. https://doi.org/10.3390/microorganisms11051145
Chicago/Turabian StyleSantos, Najara Akira Costa dos, Vanessa Rafaela de Carvalho, Jayme A. Souza-Neto, Diego Peres Alonso, Paulo Eduardo Martins Ribolla, Jansen Fernandes Medeiros, and Maisa da Silva Araujo. 2023. "Bacterial Microbiota from Lab-Reared and Field-Captured Anopheles darlingi Midgut and Salivary Gland" Microorganisms 11, no. 5: 1145. https://doi.org/10.3390/microorganisms11051145