
Apple Root Microbiome as Indicator of Plant Adaptation to Apple Replant Diseased Soils
 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Description and Sampling of Sites
2.2. DNA Extraction and Sequencing
2.3. 16S rRNA, 18S rRNA, and ITS2 Amplicon Sequence Processing
2.4. Bioinformatics and Statistical Analysis
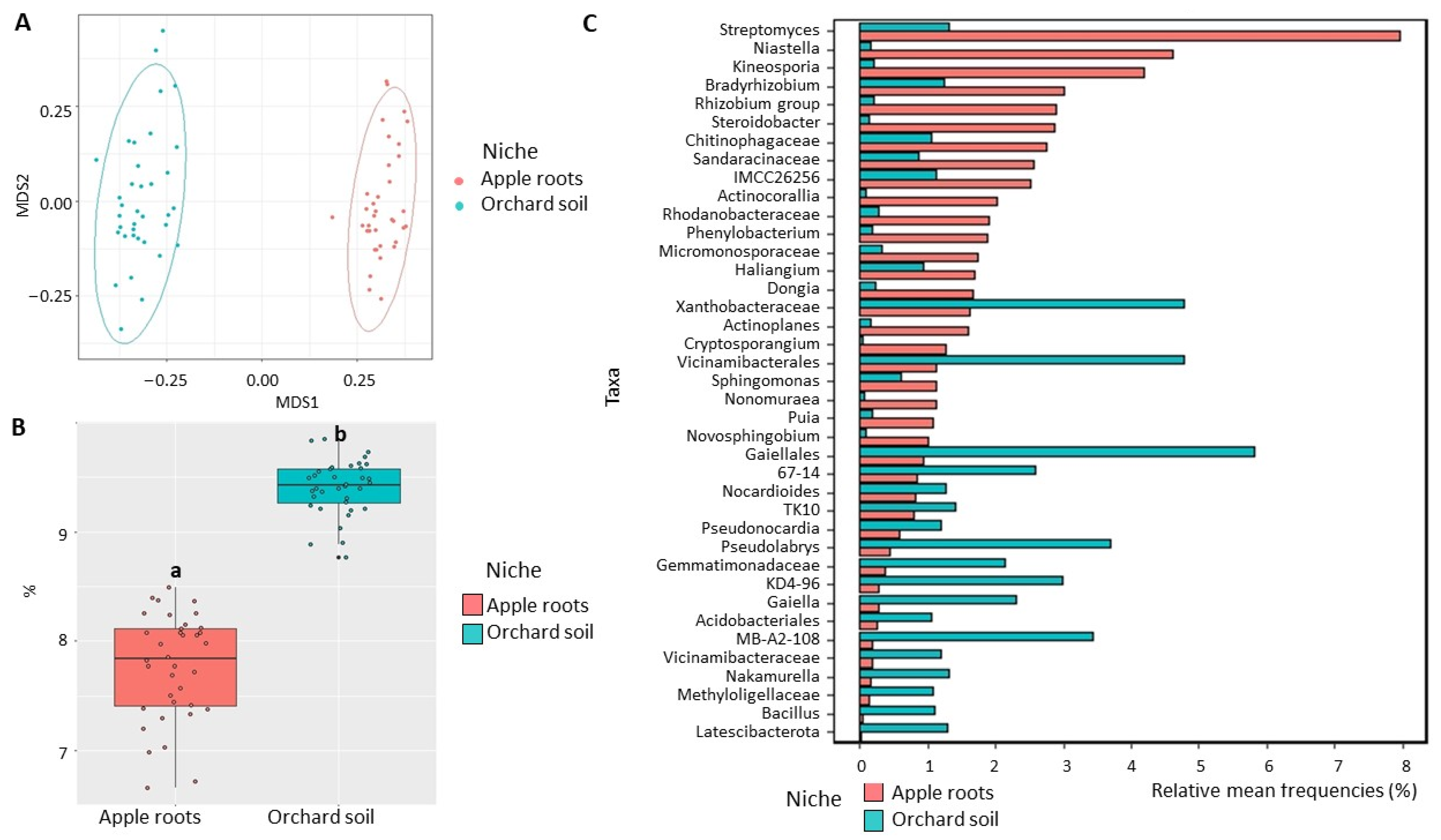
3. Results
3.1. Data Description
3.2. Effect of Soil Location on Bacterial and Eukaryotic Microbiomes
3.3. Differences between Microbial Communities of Apple Root and Orchard Soil
3.4. Variation of Soil and Apple Root Microbiome across the Orchards
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Manici, L.M.; Kelderer, M.; Franke-Whittle, I.H.; Rühmer, T.; Baab, G.; Nicoletti, F.; Caputo, F.; Topp, A.; Insam, H.; Naef, A. Relationship between Root-Endophytic Microbial Communities and Replant Disease in Specialized Apple Growing Areas in Europe. Appl. Soil Ecol. 2013, 72, 207–214. [Google Scholar] [CrossRef]
- Winkelmann, T.; Smalla, K.; Amelung, W.; Baab, G.; Grunewaldt-Stocker, G.; Kanfra, X.; Meyhofer, R.; Reim, S.; Schmitz, M.; Vetterlein, D.; et al. Apple Replant Disease: Causes and Mitigation Strategies. Curr. Issues Mol. Biol. 2019, 30, 89–106. [Google Scholar] [CrossRef]
- Wang, L.; Mazzola, M. Field Evaluation of Reduced Rate Brassicaceae Seed Meal Amendment and Rootstock Genotype on the Microbiome and Control of Apple Replant Disease. Phytopathology 2019, 109, 1378–1391. [Google Scholar] [CrossRef]
- Mazzola, M.; Manici, L.M. Apple Replant Disease: Role of Microbial Ecology in Cause and Control. Annu. Rev. Phytopathol. 2012, 50, 45–65. [Google Scholar] [CrossRef]
- Mazzola, M. Elucidation of the Microbial Complex Having a Causal Role in the Development of Apple Replant Disease in Washington. Phytopathology 1998, 88, 930–938. [Google Scholar] [CrossRef]
- Braun, P.G. The Combination of Cylindrocarpon lucidum and Pythium irregulare as a Possible Cause of Apple Replant Disease in Nova Scotia. Can. J. Plant Pathol. 1991, 13, 291–297. [Google Scholar] [CrossRef]
- Braun, P.G. Effects of Cylindrocarpon and Pythium Species on Apple Seedlings and Potential Role in Apple Replant Disease. Can. J. Plant Pathol. 1995, 17, 336–341. [Google Scholar] [CrossRef]
- Watson, T.T.; Forge, T.A.; Nelson, L.M. Pseudomonads Contribute to Regulation of Pratylenchus penetrans (Nematoda) Populations on Apple. Can. J. Microbiol. 2018, 64, 775–785. [Google Scholar] [CrossRef]
- Larousse, M.; Rancurel, C.; Syska, C.; Palero, F.; Etienne, C.; Industri, B.; Nesme, X.; Bardin, M.; Galiana, E. Tomato Root Microbiota and Phytophthora parasitica-Associated Disease. Microbiome 2017, 5, 56. [Google Scholar] [CrossRef]
- Zhang, R.; Vivanco, J.M.; Shen, Q. The Unseen Rhizosphere Root–Soil–Microbe Interactions for Crop Production. Curr. Opin. Microbiol. 2017, 37, 8–14. [Google Scholar] [CrossRef]
- Somera, T.S.; Mazzola, M. Toward a Holistic View of Orchard Ecosystem Dynamics: A Comprehensive Review of the Multiple Factors Governing Development or Suppression of Apple Replant Disease. Front. Microbiol. 2022, 13, 949404. [Google Scholar] [CrossRef]
- van der Putten, W.H.; Bardgett, R.D.; Bever, J.D.; Bezemer, T.M.; Casper, B.B.; Fukami, T.; Kardol, P.; Klironomos, J.N.; Kulmatiski, A.; Schweitzer, J.A.; et al. Plant-Soil Feedbacks: The Past, the Present and Future Challenges. J. Ecol. 2013, 101, 265–276. [Google Scholar] [CrossRef]
- Tilston, E.L.; Deakin, G.; Bennett, J.; Passey, T.; Harrison, N.; O’Brien, F.; Fernández-Fernández, F.; Xu, X. Candidate Causal Organisms for Apple Replant Disease in the United Kingdom. Phytobiomes J. 2018, 2, 261–274. [Google Scholar] [CrossRef]
- Leisso, R.; Rudell, D.; Mazzola, M. Targeted Metabolic Profiling Indicates Apple Rootstock Genotype-Specific Differences in Primary and Secondary Metabolite Production and Validate Quantitative Contribution from Vegetative Growth. Front. Plant Sci. 2018, 9, 1336. [Google Scholar] [CrossRef]
- Leisso, R.; Rudell, D.; Mazzola, M. Metabolic Composition of Apple Rootstock Rhizodeposits Differs in a Genotype-Specific Manner and Affects Growth of Subsequent Plantings. Soil Biol. Biochem. 2017, 113, 201–214. [Google Scholar] [CrossRef]
- Kanfra, X.; Liu, B.; Beerhues, L.; Sørensen, S.J.; Heuer, H. Free-Living Nematodes Together with Associated Microbes Play an Essential Role in Apple Replant Disease. Front. Plant Sci. 2018, 9, 1666. [Google Scholar] [CrossRef]
- Lucas, M.; Balbín-Suárez, A.; Smalla, K.; Vetterlein, D. Root Growth, Function and Rhizosphere Microbiome Analyses Show Local Rather than Systemic Effects in Apple Plant Response to Replant Disease Soil. PLoS ONE 2018, 13, e0204922. [Google Scholar] [CrossRef]
- Saleem, M.; Arshad, M.; Hussain, S.; Bhatti, A.S. Perspective of Plant Growth Promoting Rhizobacteria (PGPR) Containing ACC Deaminase in Stress Agriculture. J. Ind. Microbiol. Biotechnol. 2007, 34, 635–648. [Google Scholar] [CrossRef]
- Nadeem, S.M.; Ahmad, M.; Zahir, Z.A.; Javaid, A.; Ashraf, M. The Role of Mycorrhizae and Plant Growth Promoting Rhizobacteria (PGPR) in Improving Crop Productivity under Stressful Environments. Biotechnol. Adv. 2014, 32, 429–448. [Google Scholar] [CrossRef]
- Mazzola, M.; Hewavitharana, S.S.; Strauss, S.L. Brassica Seed Meal Soil Amendments Transform the Rhizosphere Microbiome and Improve Apple Production through Resistance to Pathogen Reinfestation. Phytopathology 2015, 105, 460–469. [Google Scholar] [CrossRef]
- Wei, Z.; Gu, Y.; Friman, V.-P.; Kowalchuk, G.A.; Xu, Y.; Shen, Q.; Jousset, A. Initial Soil Microbiome Composition and Functioning Predetermine Future Plant Health. Sci. Adv. 2019, 5, eaaw0759. [Google Scholar] [CrossRef]
- Yurgel, S.N.; Douglas, G.M.; Comeau, A.M.; Mammoliti, M.; Dusault, A.; Percival, D.; Langille, M.G.I. Variation in Bacterial and Eukaryotic Communities Associated with Natural and Managed Wild Blueberry Habitats. Phytobiomes J. 2017, 1, 102–113. [Google Scholar] [CrossRef]
- White, L.; Brozel, V.; Subramanian, S. Isolation of Rhizosphere Bacterial Communities from Soil. Bio-Protocol 2015, 5, e1569. [Google Scholar] [CrossRef]
- Yurgel, S.N.; Nadeem, M.; Cheema, M. Microbial Consortium Associated with Crustacean Shells Composting. Microorganisms 2022, 10, 1033. [Google Scholar] [CrossRef]
- Comeau, A.M.; Douglas, G.M.; Langille, M.G.I. Microbiome Helper: A Custom and Streamlined Workflow for Microbiome Research. mSystems 2017, 2, e00127-16. [Google Scholar] [CrossRef]
- Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A Versatile Open-Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A Fast and Accurate Illumina Paired-End ReAd MergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing Taxonomic Classification of Marker-Gene Amplicon Sequences with QIIME 2’s Q2-Feature-Classifier Plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical Analysis of Taxonomic and Functional Profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An Effective Distance Metric for Microbial Community Comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef]
- Kim, B.R.; Shin, J.; Guevarra, R.B.; Lee, J.H.; Kim, D.W.; Seol, K.H.; Lee, J.H.; Kim, H.B.; Isaacson, R.E. Deciphering Diversity Indices for a Better Understanding of Microbial Communities. J. Microbiol. Biotechnol. 2017, 27, 2089–2093. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Stevens, M.H.H.; Oksanen, M.J.; Suggests, M.A.S.S. The Vegan Package. Community Ecol. Package 2007, 10, 631–637. [Google Scholar]
- Yurgel, S.N.; Ajeethan, N.; Smertenko, A. Response of Plant-Associated Microbiome to Plant Root Colonization by Exogenous Bacterial Endophyte in Perennial Crops. Front. Microbiol. 2022, 13, 965. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2; Use R!: Cham, Switzerland; Springer International Publishing: Cham, Switzerland, 2016; ISBN 978-3-319-24275-0. [Google Scholar]
- Juurakko, C.L.; Dicenzo, G.C.; Walker, V.K. Cold Acclimation in Brachypodium Is Accompanied by Changes in Above-Ground Bacterial and Fungal Communities. Plants 2021, 10, 2824. [Google Scholar] [CrossRef]
- van Horn, C.; Somera, T.S.; Mazzola, M. Comparative Analysis of the Rhizosphere and Endophytic Microbiomes across Apple Rootstock Genotypes in Replant Orchard Soils. Phytobiomes J. 2021, 5, 231–243. [Google Scholar] [CrossRef]
- Deakin, G.; Tilston, E.L.; Bennett, J.; Passey, T.; Harrison, N.; Fernandez-Fernandez, F.; Xu, X. Spatial Structuring of Soil Microbial Communities in Commercial Apple Orchards. Appl. Soil Ecol. 2018, 130, 1–12. [Google Scholar] [CrossRef]
- Deakin, G.; Fernandez-Fernandez, F.; Bennett, J.; Passey, T.; Harrison, N.; Tilston, E.L.; Xu, X. The Effect of Rotating Apple Rootstock Genotypes on Apple Replant Disease and Rhizosphere Microbiome. Phytobiomes J. 2019, 3, 273–285. [Google Scholar] [CrossRef]
- Wicaksono, W.A.; Buko, A.; Kusstatscher, P.; Cernava, T.; Sinkkonen, A.; Laitinen, O.H.; Virtanen, S.M.; Hyoty, H.; Berg, G. Impact of Cultivation and Origin on the Fruit Microbiome of Apples and Blueberries and Implications for the Exposome. Microb. Ecol. 2022. [Google Scholar] [CrossRef]
- Yurgel, S.N.; Douglas, G.M.; Dusault, A.; Percival, D.; Langille, M.G.I. Dissecting Community Structure in Wild Blueberry Root and Soil Microbiome. Front. Microbiol. 2018, 9, 1187. [Google Scholar] [CrossRef]
- Eisenhauer, N.; Beler, H.; Engels, C.; Gleixner, G.; Habekost, M.; Milcu, A.; Partsch, S.; Sabais, A.C.W.; Scherber, C.; Steinbeiss, S.; et al. Plant Diversity Effects on Soil Microorganisms Support the Singular Hypothesis. Ecology 2010, 91, 485–496. [Google Scholar] [CrossRef]
- Riahi, H.S.; Heidarieh, P.; Fatahi-Bafghi, M. Genus Pseudonocardia: What We Know about Its Biological Properties, Abilities and Current Application in Biotechnology. J. Appl. Microbiol. 2022, 132, 890–906. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.R.; Mustafa, A.; Hyder, S.; Valipour, M.; Rizvi, Z.F.; Gondal, A.S.; Yousuf, Z.; Iqbal, R.; Daraz, U. Bacillus spp. as Bioagents: Uses and Application for Sustainable Agriculture. Biology 2022, 11, 1763. [Google Scholar] [CrossRef] [PubMed]
- Dobrzynski, J.; Jakubowska, Z.; Dybek, B. Potential of Bacillus pumilus to Directly Promote Plant Growth. Front. Microbiol. 2022, 13, 1069053. [Google Scholar] [CrossRef] [PubMed]
- Boukhatem, Z.F.; Merabet, C.; Tsaki, H. Plant Growth Promoting Actinobacteria, the Most Promising Candidates as Bioinoculants? Front. Agron. 2022, 4, 849911. [Google Scholar] [CrossRef]
- Coombs, J.T.; Michelsen, P.P.; Franco, C.M.M. Evaluation of Endophytic Actinobacteria as Antagonists of Gaeumannomyces graminis var. tritici in Wheat. Biol. Control 2004, 29, 359–366. [Google Scholar] [CrossRef]
- Fiorentino, N.; Ventorino, V.; Woo, S.L.; Pepe, O.; de Rosa, A.; Gioia, L.; Romano, I.; Lombardi, N.; Napolitano, M.; Colla, G.; et al. Trichoderma-Based Biostimulants Modulate Rhizosphere Microbial Populations and Improve N Uptake Efficiency, Yield, and Nutritional Quality of Leafy Vegetables. Front. Plant Sci. 2018, 9, 743. [Google Scholar] [CrossRef]
- Lopez-Bucio, J.; Pelagio-Flores, R.; Herrera-Estrella, A. Trichoderma as Biostimulant: Exploiting the Multilevel Properties of a Plant Beneficial Fungus. Sci. Hortic. 2015, 196, 109–123. [Google Scholar] [CrossRef]
- Howell, C.R. Mechanisms Employed by Trichoderma Species in the Biological Control of Plant Diseases: The History and Evolution of Current Concepts. Plant Dis. 2003, 87, 4–10. [Google Scholar] [CrossRef]
- Pedersen, A.L.; Nybroe, O.; Winding, A.; Ekelund, F.; Bjornlund, L. Bacterial Feeders, the Nematode Caenorhabditis elegans and the Flagellate Cercomonas longicauda, Have Different Effects on Outcome of Competition among the Pseudomonas Biocontrol Strains CHA0 and DSS73. Microb. Ecol. 2009, 57, 501–509. [Google Scholar] [CrossRef]
- Ronn, R.; McCaig, A.E.; Griffiths, B.S.; Prosser, J.I. Impact of Protozoan Grazing on Bacterial Community Structure in Soil Microcosms. Appl. Environ. Microbiol. 2002, 68, 6094–6105. [Google Scholar] [CrossRef]
- Mattison, R.G.; Harayama, S. The Predatory Soil Flagellate Heteromita globosa Stimulates Toluene Biodegradation by a Pseudomonas sp. FEMS Microbiol. Lett. 2001, 194, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Brunner-Mendoza, C.; Reyes-Montes, M.d.R.; Moonjely, S.; Bidochka, M.J.; Toriello, C. A Review on the Genus Metarhizium as an Entomopathogenic Microbial Biocontrol Agent with Emphasis on Its Use and Utility in Mexico. Biocontrol Sci. Technol. 2019, 29, 83–102. [Google Scholar] [CrossRef]
- Siqueira, A.C.O.; Mascarin, G.M.; Gonçalves, C.R.N.C.B.; Marcon, J.; Quecine, M.C.; Figueira, A.; Delalibera, Í. Multi-Trait Biochemical Features of Metarhizium Species and Their Activities That Stimulate the Growth of Tomato Plants. Front. Sustain. Food Syst. 2020, 4, 137. [Google Scholar] [CrossRef]
- Tapia-Vazquez, I.; Sanchez-Cruz, R.; Arroyo-Dominguez, M.; Lira-Ruan, V.; Sanchez-Reyes, A.; del Rayo Sanchez-Carbente, M.; Padilla-Chacon, D.; Batista-Garcia, R.A.; Folch-Mallol, J.L. Isolation and Characterization of Psychrophilic and Psychrotolerant Plant-Growth Promoting Microorganisms from a High-Altitude Volcano Crater in Mexico. Microbiol. Res. 2020, 232, 126394. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Saltzgiver, M. A Systematic Analysis of Apple Root Resistance Traits to Pythium ultimum Infection and the Underpinned Molecular Regulations of Defense Activation. Hortic. Res. 2020, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.N.; Jiang, W.T.; Zhang, R.; Chen, R.; Chen, X.S.; Yin, C.M.; Mao, Z.Q. Discovery of Fusarium proliferatum f. sp. Malus domestica Causing Apple Replant Disease in China. Plant Dis. 2022, 106, 2958–2966. [Google Scholar] [CrossRef]
- Kanfra, X.; Wrede, A.; Moll, J.; Heuer, H. Nematode–Microbe Complexes in Soils Replanted with Apple. Microorganisms 2022, 10, 157. [Google Scholar] [CrossRef]
- Hoestra, H. Replant Diseases of Apple in the Netherlands; Wageningen University and Research: Wageningen, The Netherlands, 1968; Available online: https://edepot.wur.nl/191751 (accessed on 23 November 2022).
- Mao, Y.; Li, X.; Smyth, E.M.; Yannarell, A.C.; Mackie, R.I. Enrichment of Specific Bacterial and Eukaryotic Microbes in the Rhizosphere of Switchgrass (Panicum virgatum L.) through Root Exudates. Environ. Microbiol. Rep. 2014, 6, 293–306. [Google Scholar] [CrossRef]
- Hargreaves, S.K.; Williams, R.J.; Hofmockel, K.S. Environmental Filtering of Microbial Communities in Agricultural Soil Shifts with Crop Growth. PLoS ONE 2015, 10, e0134345. [Google Scholar] [CrossRef]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant–Microbiome Interactions: From Community Assembly to Plant Health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an Ecological Classification of Soil Bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.; di Lonardo, D.P.; Bodelier, P.L.E. Revisiting Life Strategy Concepts in Environmental Microbial Ecology. FEMS Microbiol. Ecol. 2017, 93, fix006. [Google Scholar] [CrossRef] [PubMed]
- Mącik, M.; Gryta, A.; Frąc, M. Biofertilizers in Agriculture: An Overview on Concepts, Strategies and Effects on Soil Microorganisms. In Advances in Agronomy; Academic Press Inc.: Cambridge, MA, USA, 2020; Volume 162, pp. 31–87. ISBN 9780128207673. [Google Scholar]
- Asaf, S.; Numan, M.; Khan, A.L.; Al-Harrasi, A. Sphingomonas: From Diversity and Genomics to Functional Role in Environmental Remediation and Plant Growth. Crit. Rev. Biotechnol. 2020, 40, 138–152. [Google Scholar] [CrossRef] [PubMed]
- van Gerrewey, T.; El-Nakhel, C.; de Pascale, S.; de Paepe, J.; Clauwaert, P.; Kerckhof, F.M.; Boon, N.; Geelen, D. Root-Associated Bacterial Community Shifts in Hydroponic Lettuce Cultured with Urine-Derived Fertilizer. Microorganisms 2021, 9, 1326. [Google Scholar] [CrossRef]
- Vurukonda, S.S.K.P.; Giovanardi, D.; Stefani, E. Plant Growth Promoting and Biocontrol Activity of Streptomyces spp. as Endophytes. Int. J. Mol. Sci. 2018, 19, 952. [Google Scholar] [CrossRef] [PubMed]
- Malicka, M.; Magurno, F.; Piotrowska-Seget, Z. Plant Association with Dark Septate Endophytes: When the Going Gets Tough (and Stressful), the Tough Fungi Get Going. Chemosphere 2022, 302, 134830. [Google Scholar] [CrossRef] [PubMed]
- Popp, C.; Wamhoff, D.; Winkelmann, T.; Maiss, E.; Grunewaldt-Stöcker, G. Molecular Identification of Nectriaceae in Infections of Apple Replant Disease Affected Roots Collected by Harris Uni-Core Punching or Laser Microdissection. J. Plant Dis. Prot. 2020, 127, 571–582. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Amplicon | Parameter | R2 |
|---|---|---|
| 16S rRNA | ||
| Apple root/Orchard soil | Niche | 0.305 *** |
| Location | 0.157 *** | |
| Niche × Location | 0.104 *** | |
| Orchard/Uncultivated soil | Niche | 0.288 *** |
| Apple root | Location | 0.358 *** |
| Orchard soil | Location | 0.399 *** |
| ITS | ||
| Apple root/Orchard soil | Niche | 0.389 *** |
| Location | 0.142 *** | |
| Niche × Location | 0.082 ** | |
| Apple root | Location | 0.309 *** |
| Orchard soil | Location | 0.424 *** |
| 18S rRNA | ||
| Orchard/Uncultivated soil | Niche | 0.304 *** |
| Orchard soil | Location | 0.341 *** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ajeethan, N.; Ali, S.; Fuller, K.D.; Abbey, L.; Yurgel, S.N. Apple Root Microbiome as Indicator of Plant Adaptation to Apple Replant Diseased Soils. Microorganisms 2023, 11, 1372. https://doi.org/10.3390/microorganisms11061372
Ajeethan N, Ali S, Fuller KD, Abbey L, Yurgel SN. Apple Root Microbiome as Indicator of Plant Adaptation to Apple Replant Diseased Soils. Microorganisms. 2023; 11(6):1372. https://doi.org/10.3390/microorganisms11061372
Chicago/Turabian StyleAjeethan, Nivethika, Shawkat Ali, Keith D. Fuller, Lord Abbey, and Svetlana N. Yurgel. 2023. "Apple Root Microbiome as Indicator of Plant Adaptation to Apple Replant Diseased Soils" Microorganisms 11, no. 6: 1372. https://doi.org/10.3390/microorganisms11061372
APA StyleAjeethan, N., Ali, S., Fuller, K. D., Abbey, L., & Yurgel, S. N. (2023). Apple Root Microbiome as Indicator of Plant Adaptation to Apple Replant Diseased Soils. Microorganisms, 11(6), 1372. https://doi.org/10.3390/microorganisms11061372