
Comparative Genomic Analysis of ST131 Subclade C2 of ESBL-Producing E. coli Isolates from Patients with Recurrent and Sporadic Urinary Tract Infections

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Isolates
2.2. Whole Genome Sequencing and Assembly
2.3. Genomic Characterization of the Isolates
2.4. Pan and Core Genome Analysis
3. Results and Discussion
3.1. Genome-Based Typing
3.2. Virulence and Antibiotic Resistance Determinants
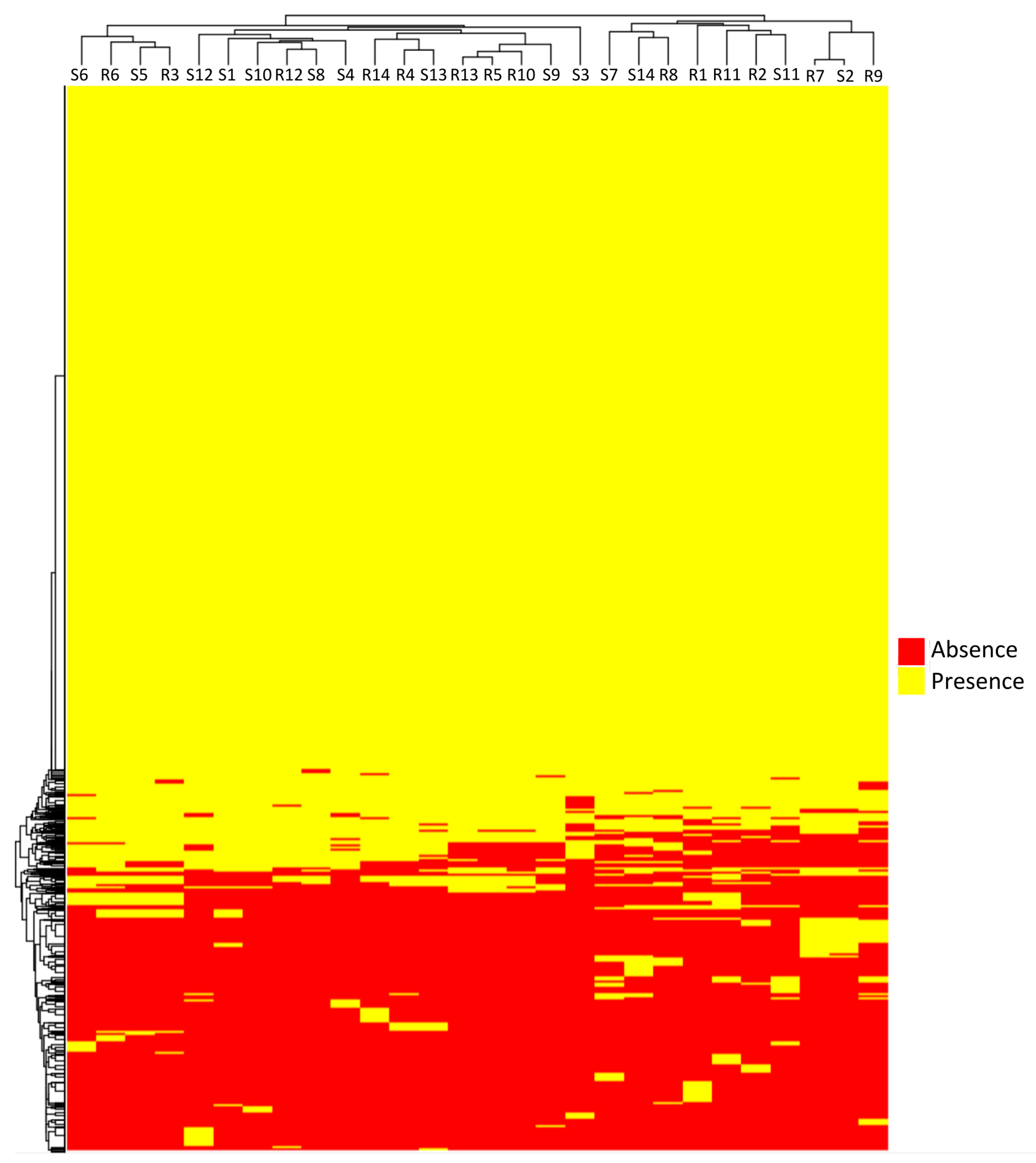
3.3. Pangenome Analysis
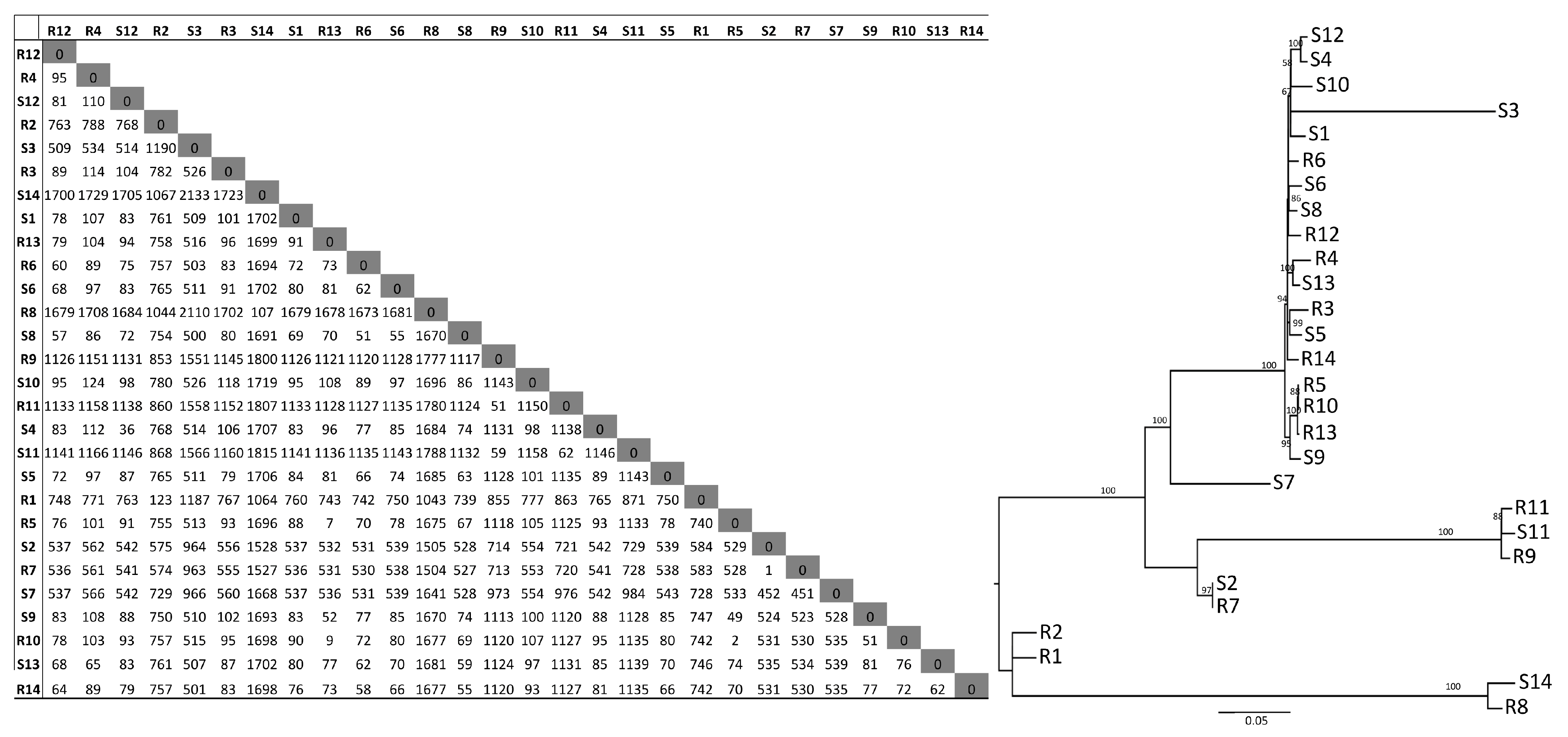
3.4. SNP Analysis
3.5. Limitations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hooton, T.M. Clinical practice. Uncomplicated urinary tract infection. N. Engl. J. Med. 2012, 366, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Russo, T.A.; Johnson, J.R. Medical and economic impact of extraintestinal infections due to Escherichia coli: Focus on an increasingly important endemic problem. Microbes Infect. 2003, 5, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Foxman, B.; Gillespie, B.; Koopman, J.; Zhang, L.; Palin, K.; Tallman, P.; Marsh, J.V.; Spear, S.; Sobel, J.D.; Marty, M.J.; et al. Risk factors for second urinary tract infection among college women. Am. J. Epidemiol. 2000, 151, 1194–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindblom, A.; Karami, N.; Magnusson, T.; Ahren, C. Subsequent infection with extended-spectrum beta-lactamase-producing Enterobacteriaceae in patients with prior infection or fecal colonization. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Peirano, G.; Pitout, J.D. Molecular epidemiology of Escherichia coli producing CTX-M beta-lactamases: The worldwide emergence of clone ST131 O25, H4. Int. J. Antimicrob. Agents 2010, 35, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Peirano, G.; Lynch, T.; Matsumara, Y.; Nobrega, D.; Finn, T.J.; Devinney, R.; Pitout, J.D. Trends in Population Dynamics of Escherichia coli Sequence Type 131, Calgary, Alberta, Canada, 2006–2016(1). Emerg. Infect. Dis. 2020, 26, 2907–2915. [Google Scholar] [CrossRef]
- Pitout, J.D.D.; Finn, T.J. The evolutionary puzzle of Escherichia coli ST131. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2020, 81, 104265. [Google Scholar] [CrossRef]
- Petty, N.K.; Ben Zakour, N.L.; Stanton-Cook, M.; Skippington, E.; Totsika, M.; Forde, B.M.; Phan, M.-D.; Moriel, D.G.; Peters, K.M.; Davies, M.; et al. Global dissemination of a multidrug resistant Escherichia coli clone. Proc. Natl. Acad. Sci. USA 2014, 111, 5694–5699. [Google Scholar] [CrossRef] [Green Version]
- Biggel, M.; Moons, P.; Nguyen, M.N.; Goossens, H.; Van Puyvelde, S. Convergence of virulence and antimicrobial resistance in increasingly prevalent Escherichia coli ST131 papGII+ sublineages. Commun. Biol. 2022, 5, 752. [Google Scholar] [CrossRef]
- Pajand, O.; Rahimi, H.; Darabi, N.; Roudi, S.; Ghassemi, K.; Aarestrup, F.M.; Leekitcharoenphon, P. Arrangements of Mobile Genetic Elements among Virotype E Subpopulation of Escherichia coli Sequence Type 131 Strains with High Antimicrobial Resistance and Virulence Gene Content. mSphere 2021, 6, e0055021. [Google Scholar] [CrossRef]
- Hojabri, Z.; Darabi, N.; Mirmohammadkhani, M.; Rahimi, H.; Hemmati, R.; Saeedi, Z.; Roustaee, K.; Leekitcharoenphon, P.; Pajand, O.; Aarestrup, F.M. Expansion of a Subset Within the C2 Subclade of Escherichia coli Sequence Type 131 (ST131) Is Driving the Increasing Rates of Aminoglycoside Resistance. Open Forum Infect. Dis. 2020, 7, ofaa410. [Google Scholar] [CrossRef]
- Karami, N.; Lindblom, A.; Yazdanshenas, S.; Linden, V.; Ahren, C. Recurrence of urinary tract infections with extended-spectrum beta-lactamase-producing Escherichia coli caused by homologous strains among which clone ST131-O25b is dominant. J. Glob. Antimicrob. Resist. 2020, 22, 126–132. [Google Scholar] [CrossRef]
- Lindblom, A.; Kiszakiewicz, C.; Kristiansson, E.; Yazdanshenas, S.; Kamenska, N.; Karami, N.; Åhrén, C. The impact of the ST131 clone on recurrent ESBL-producing E. coli urinary tract infection: A prospective comparative study. Sci. Rep. 2022, 12, 10048. [Google Scholar] [CrossRef]
- Salvà-Serra, F.; Svensson-Stadler, L.; Busquets, A.; Jaén-Luchoro, D.; Karlsson, R.; Moore, E.R.; Gomila, M. A protocol for extraction and purification of high-quality and quantity bacterial DNA applicable for genome sequencing: A modified version of the Marmur procedure. Protoc. Exch. 2018. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [Green Version]
- Tatusova, T.; Dicuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Bessonov, K.; Laing, C.; Robertson, J.; Yong, I.; Ziebell, K.; Gannon, V.P.J.; Nichani, A.; Arya, G.; Nash, J.H.E.; Christianson, S. ECTyper: In silico Escherichia coli serotype and species prediction from raw and assembled whole-genome sequence data. Microb. Genom. 2021, 7, 000728. [Google Scholar] [CrossRef]
- Waters, N.R.; Abram, F.; Brennan, F.; Holmes, A.; Pritchard, L. Easy phylotyping of Escherichia coli via the EzClermont web app and command-line tool. Access Microbiol. 2020, 2, acmi000143. [Google Scholar] [CrossRef]
- Roer, L.; Tchesnokova, V.; Allesoe, R.; Muradova, M.; Chattopadhyay, S.; Ahrenfeldt, J.; Thomsen, M.C.; Lund, O.; Hansen, F.; Hammerum, A.M.; et al. Development of a Web Tool for Escherichia coli Subtyping Based on fimH Alleles. J. Clin. Microbiol. 2017, 55, 2538–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcarthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef] [PubMed]
- Gonzales-Siles, L.; Karlsson, R.; Schmidt, P.; Salva-Serra, F.; Jaen-Luchoro, D.; Skovbjerg, S.; Moore, E.R.; Gomila, M. A Pangenome Approach for Discerning Species-Unique Gene Markers for Identifications of Streptococcus pneumoniae and Streptococcus pseudopneumoniae. Front. Cell. Infect. Microbiol. 2020, 10, 222. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Clausen, P.; Aarestrup, F.M.; Lund, O. Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinform. 2018, 19, 307. [Google Scholar] [CrossRef]
- Zhou, Z.; Alikhan, N.F.; Mohamed, K.; Fan, Y.; Agama Study, G.; Achtman, M. The EnteroBase user’s guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome Res. 2020, 30, 138–152. [Google Scholar] [CrossRef] [Green Version]
- Brynildsrud, O.; Bohlin, J.; Scheffer, L.; Eldholm, V. Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 2016, 17, 238. [Google Scholar] [CrossRef] [Green Version]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [Green Version]
- Blanco, J.; Mora, A.; Mamani, R.; Lopez, C.; Blanco, M.; Dahbi, G.; Herrera, A.; Marzoa, J.; Fernández, V.; De La Cruz, F.; et al. Four main virotypes among extended-spectrum-beta-lactamase-producing isolates of Escherichia coli O25b:H4-B2-ST131: Bacterial, epidemiological, and clinical characteristics. J. Clin. Microbiol. 2013, 51, 3358–3367. [Google Scholar] [CrossRef] [Green Version]
- Forde, B.M.; Roberts, L.W.; Phan, M.-D.; Peters, K.M.; Fleming, B.A.; Russell, C.W.; Lenherr, S.M.; Myers, J.B.; Barker, A.P.; Fisher, M.A.; et al. Population dynamics of an Escherichia coli ST131 lineage during recurrent urinary tract infection. Nat. Commun. 2019, 10, 3643. [Google Scholar] [CrossRef] [Green Version]
- Soto, S.M.; Smithson, A.; Martinez, J.A.; Horcajada, J.P.; Mensa, J.; Vila, J. Biofilm formation in uropathogenic Escherichia coli strains: Relationship with prostatitis, urovirulence factors and antimicrobial resistance. J. Urol. 2007, 177, 365–368. [Google Scholar] [CrossRef]
- Thanert, R.; Reske, K.A.; Hink, T.; Wallace, M.A.; Wang, B.; Schwartz, D.J.; Seiler, S.; Cass, C.; Burnham, C.-A.D.; Dubberke, E.R.; et al. Comparative Genomics of Antibiotic-Resistant Uropathogens Implicates Three Routes for Recurrence of Urinary Tract Infections. mBio 2019, 10, e01977-19. [Google Scholar] [CrossRef] [Green Version]
- Moreno, E.; Andreu, A.; Pigrau, C.; Kuskowski, M.A.; Johnson, J.R.; Prats, G. Relationship between Escherichia coli strains causing acute cystitis in women and the fecal E. coli population of the host. J. Clin. Microbiol. 2008, 46, 2529–2534. [Google Scholar] [CrossRef] [Green Version]
- Kerrn, M.B.; Struve, C.; Blom, J.; Frimodt-Moller, N.; Krogfelt, K.A. Intracellular persistence of Escherichia coli in urinary bladders from mecillinam-treated mice. J. Antimicrob. Chemother. 2005, 55, 383–386. [Google Scholar] [CrossRef]
- Johnson, J.R.; O’bryan, T.T.; Delavari, P.; Kuskowski, M.; Stapleton, A.; Carlino, U.; Russo, T.A. Clonal relationships and extended virulence genotypes among Escherichia coli isolates from women with a first or recurrent episode of cystitis. J. Infect. Dis. 2001, 183, 1508–1517. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, K.L.; Stegger, M.; Kiil, K.; Lilje, B.; Ejrnaes, K.; Leihof, R.F.; Skjøt-Rasmussen, L.; Godfrey, P.; Monsen, T.; Ferry, S.; et al. Escherichia coli Causing Recurrent Urinary Tract Infections: Comparison to Non-Recurrent Isolates and Genomic Adaptation in Recurrent Infections. Microorganisms 2021, 9, 1416. [Google Scholar] [CrossRef]
- Ejrnaes, K.; Stegger, M.; Reisner, A.; Ferry, S.; Monsen, T.; Holm, S.E.; Lundgren, B.; Frimodt-Møller, N. Characteristics of Escherichia coli causing persistence or relapse of urinary tract infections: Phylogenetic groups, virulence factors and biofilm formation. Virulence 2011, 2, 528–537. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.R.; Porter, S.; Johnston, B.; Kuskowski, M.A.; Spurbeck, R.R.; Mobley, H.L.; Williamson, D.A. Host Characteristics and Bacterial Traits Predict Experimental Virulence for Escherichia coli Bloodstream Isolates from Patients with Urosepsis. Open Forum Infect. Dis. 2015, 2, ofv083. [Google Scholar] [CrossRef]
- Duprilot, M.; Baron, A.; Blanquart, F.; Dion, S.; Pouget, C.; Letteron, P.; Flament-Simon, S.-C.; Clermont, O.; Denamur, E.; Nicolas-Chanoine, M.-H. Success of Escherichia coli O25b:H4 Sequence Type 131 Clade C Associated with a Decrease in Virulence. Infect. Immun. 2020, 88, e00576-20. [Google Scholar] [CrossRef]
- Hertz, F.B.; Marvig, R.L.; Frimodt-Moller, N.; Nielsen, K.L. In vitro Relative Fitness, in vivo Intestinal Colonization and Genomic Differences of Escherichia coli of ST131 Carrying bla (CTX-M-15). Front. Microbiol. 2021, 12, 798473. [Google Scholar] [CrossRef] [PubMed]
- McNally, A.; Kallonen, T.; Connor, C.; Abudahab, K.; Aanensen, D.M.; Horner, C.; Peacock, S.J.; Parkhill, J.; Croucher, N.J.; Corander, J. Diversification of Colonization Factors in a Multidrug-Resistant Escherichia coli Lineage Evolving under Negative Frequency-Dependent Selection. mBio 2019, 10, e00644-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Reid, C.J.; Kudinha, T.; Jarocki, V.M.; Djordjevic, S.P. Genomic analysis of trimethoprim-resistant extraintestinal pathogenic Escherichia coli and recurrent urinary tract infections. Microb. Genom. 2020, 6, e000475. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.L.; Ding, Y.; Apisarnthanarak, A.; Kalimuddin, S.; Archuleta, S.; Omar, S.F.S.; De, P.P.; Koh, T.H.; Chew, K.L.; Atiya, N.; et al. The higher prevalence of extended spectrum beta-lactamases among Escherichia coli ST131 in Southeast Asia is driven by expansion of a single, locally prevalent subclone. Sci. Rep. 2019, 9, 13245. [Google Scholar] [CrossRef] [Green Version]
- Nobrega, D.; Peirano, G.; Lynch, T.; Finn, T.J.; Devinney, R.; Pitout, J.D.D. Spatial distribution of Escherichia coli ST131 C subclades in a centralized Canadian urban region. J. Antimicrob. Chemother. 2021, 76, 1135–1139. [Google Scholar] [CrossRef]
- Jaen-Luchoro, D.; Karlsson, R.; Busquets, A.; Pineiro-Iglesias, B.; Karami, N.; Marathe, N.P.; Moore, E.R. Knockout of Targeted Plasmid-Borne beta-Lactamase Genes in an Extended-Spectrum-beta-Lactamase-Producing Escherichia coli Strain: Impact on Resistance and Proteomic Profile. Microbiol. Spectr. 2023, 11, e0386722. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient and Isolates ID | Sex | Age (Year) | HC/PC b | Date of Isolation | Phenotypic Resistance to | Days to First Recurrence | Virotype d | ||
|---|---|---|---|---|---|---|---|---|---|
| CIP c | TOB c | TMP c | |||||||
| Recurrent UTI | |||||||||
| R1 | M | 77 | PC | Oct. 2017 | R | S | R | 40 | C |
| R2 | M | 80 | HC | Oct. 2017 | R | R | R | 68 | C |
| R3a | M | 51 | HC | Oct. 2017 | R | R | S | 42 | C |
| R4 | M | 59 | PC | Nov. 2017 | R | R | S | 158 | C |
| R5 | F | 88 | HC | Nov. 2017 | R | R | R | 58 | C |
| R6 a | F | 21 | HC | Dec. 2017 | R | R | S | 78 | C |
| R7 | M | 86 | HC | Feb. 2018 | R | R | S | 106 | A |
| R8 | F | 65 | HC | Mar. 2018 | R | R | S | 41 | C |
| R9 a | F | 59 | HC | May 2018 | R | S | S | 33 | A |
| R10 | M | 78 | HC | Jun. 2018 | R | R | S | 31 | C |
| R11 a | F | 89 | HC | Jun. 2018 | R | R | S | 62 | A |
| R12 | F | 34 | HC | Aug. 2018 | R | R | R | 73 | C |
| R13 a | F | 80 | HC | Sep. 2018 | R | R | R | 146 | C |
| R14 a | F | 74 | PC | Sep. 2018 | R | R | R | 54 | C |
| Sporadic UTI | |||||||||
| S1 | M | 73 | HC | Nov. 2017 | R | R | R | C | |
| S2 | F | 78 | PC | Dec. 2017 | R | R | S | C | |
| S3 | M | 74 | PC | Jan. 2018 | R | R | S | None | |
| S4 | M | 74 | HC | Jan. 2018 | R | R | R | C | |
| S5 | M | 51 | HC | Jan. 2018 | R | R | R | C | |
| S6 | F | 68 | PC | Feb. 2018 | R | R | S | C | |
| S7 | F | 85 | PC | Mar. 2018 | R | R | R | C | |
| S8 | F | 25 | HC | May 2018 | R | R | S | C | |
| S9 | F | 69 | PC | May 2018 | R | R | R | C | |
| S10 | F | 47 | HC | May 2018 | R | R | R | C | |
| S11 | F | 18 | HC | Jul. 2018 | R | R | R | C | |
| S12 | M | 84 | HC | Aug. 2018 | R | R | R | C | |
| S13 | F | 86 | HC | Sep. 2018 | R | R | S | C | |
| S14 | M | 63 | PC | Sep. 2018 | R | R | R | C | |
| Adhesins/Fimbria | Toxins | T2SS | Hemolysins | Others | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Isolate | afaA,B-I,C-I,D | afaE-I | papCDFGHJK | papE | fimB | senB | cnf1 | gspCDEFGHIJK | hlyABD | hlyC | draP | astA |
| R1 | . | . | Y | . | . | . | . | Y | . | . | . | . |
| R2 | . | . | . | . | . | . | . | Y | . | . | . | . |
| R3 | . | . | Y | . | . | Y | Y | . | Y | Y | . | . |
| R4 | . | . | Y | . | . | . | Y | . | Y | Y | . | . |
| R5 | . | . | Y | . | . | Y | . | . | . | . | . | . |
| R6 | . | . | Y | . | . | Y | Y | . | Y | Y | . | . |
| R7 | Y | Y | . | . | . | Y | . | . | . | . | Y | . |
| R8 | . | . | . | . | Y | Y1 | . | Y | . | . | . | . |
| R9 | Y | . | . | . | . | . | . | Y | . | . | Y | . |
| R10 | . | . | Y | . | . | . | . | . | . | . | . | . |
| R11 | Y | . | . | . | . | . | . | Y | . | . | Y | . |
| R12 | . | . | Y | . | . | . | Y | . | Y | Y | . | . |
| R13 | . | . | Y | . | . | Y | . | . | . | . | . | . |
| R14 | . | . | Y | . | . | . | Y | . | Y | Y | . | . |
| S1 | . | . | Y | . | . | Y | Y | . | Y | Y | . | . |
| S2 | Y | Y | . | . | . | Y | . | . | . | . | Y | . |
| S3 | . | . | Y | . | . | . | Y | . | Y | Y | . | . |
| S4 | . | . | Y | . | . | . | Y | . | Y | Y | . | . |
| S5 | . | . | Y | . | . | Y | Y | . | Y | Y | . | . |
| S6 | . | . | Y | . | . | . | Y | . | Y | Y | . | . |
| S7 | Y | . | . | . | . | . | . | . | . | . | Y | . |
| S8 | . | . | Y | . | . | Y | Y | . | Y | Y | . | . |
| S9 | . | . | Y | . | . | Y | . | . | . | . | . | . |
| S10 | . | . | Y | . | . | Y | Y | . | Y | Y | . | Y |
| S11 | Y | . | . | . | . | . | . | Y | . | . | Y | . |
| S12 | . | . | Y | . | . | . | Y | . | Y | Y | . | . |
| S13 | . | . | Y | . | . | . | Y | . | Y | . | . | . |
| S14 | . | . | Y | Y | Y | Y | . | Y | Y | Y | . | . |
| Aminoglycosides | Macrolides | β-lactams | Diaminopyrimidines | Sulfonamides | Tetracyclins | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Isolate | AAC(3)-IIe | AAC(6′)-Ib-cr | APH(3″)-Ib | APH(6)-Id | aadA2 | aadA5 | mphA | OXA-1 | TEM-1 | dfrA12 | dfrA17 | sul1 | sul2 | tet(A) |
| R1 | . | . | . | . | . | . | . | . | . | . | . | . | . | . |
| R2 | . | Y | . | . | Y | . | Y | Y | . | Y | . | Y | . | Y |
| R3 | Y | Y | . | . | . | . | . | Y | . | . | . | . | . | . |
| R4 | Y | Y | . | . | . | . | . | Y | . | . | . | . | . | . |
| R5 | . | Y | . | . | . | Y | Y | Y | . | . | Y | Y | . | Y |
| R6 | . | Y | . | . | . | . | . | Y | . | . | . | . | . | Y |
| R7 | . | Y | Y | Y | . | . | . | Y | . | . | . | . | Y | Y |
| R8 | Y | Y | . | . | . | . | . | Y | . | . | . | Y | . | . |
| R9 | . | . | . | . | . | . | . | . | . | . | . | . | . | . |
| R10 | . | Y | . | . | . | . | . | Y | . | . | . | . | . | Y |
| R11 | . | Y | . | . | . | . | . | Y | . | . | . | . | . | Y |
| R12 | Y | Y | . | . | . | . | . | Y | . | . | . | . | . | . |
| R13 | . | Y | . | . | . | Y | Y | Y | . | . | Y | Y | . | Y |
| R14 | Y | Y | . | . | Y | . | Y | Y | . | Y | . | Y | . | Y |
| S1 | Y | Y | . | . | . | Y | Y | Y | . | . | Y | Y | . | Y |
| S2 | . | Y | Y | Y | . | . | . | Y | . | . | . | . | Y | Y |
| S3 | Y | Y | . | . | . | . | . | Y | . | . | . | . | . | . |
| S4 | Y | Y | . | . | . | . | Y | Y | . | . | Y | Y | . | Y |
| S5 | Y | Y | . | . | . | Y | Y | Y | . | . | Y | Y | . | Y |
| S6 | . | Y | . | . | . | . | . | Y | . | . | . | . | . | . |
| S7 | Y | Y | Y | Y | . | Y | Y | Y | Y | . | Y | Y | Y | Y |
| S8 | Y | Y | . | . | . | . | . | Y | . | . | . | . | . | Y |
| S9 | . | Y | . | . | . | Y | Y | Y | . | . | Y | Y | . | Y |
| S10 | Y | Y | . | . | . | Y | Y | Y | . | . | Y | Y | . | . |
| S11 | . | Y | . | . | . | Y | Y | Y | . | . | Y | Y | . | . |
| S12 | Y | Y | . | . | . | Y | Y | Y | . | . | Y | Y | . | Y |
| S13 | Y | Y | . | . | . | . | . | Y | . | . | . | . | . | . |
| S14 | . | . | Y | Y | . | Y | Y | . | Y | . | Y | Y | Y | Y |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaén-Luchoro, D.; Kahnamouei, A.; Yazdanshenas, S.; Lindblom, A.; Samuelsson, E.; Åhrén, C.; Karami, N. Comparative Genomic Analysis of ST131 Subclade C2 of ESBL-Producing E. coli Isolates from Patients with Recurrent and Sporadic Urinary Tract Infections. Microorganisms 2023, 11, 1622. https://doi.org/10.3390/microorganisms11071622
Jaén-Luchoro D, Kahnamouei A, Yazdanshenas S, Lindblom A, Samuelsson E, Åhrén C, Karami N. Comparative Genomic Analysis of ST131 Subclade C2 of ESBL-Producing E. coli Isolates from Patients with Recurrent and Sporadic Urinary Tract Infections. Microorganisms. 2023; 11(7):1622. https://doi.org/10.3390/microorganisms11071622
Chicago/Turabian StyleJaén-Luchoro, Daniel, Arezou Kahnamouei, Shora Yazdanshenas, Anna Lindblom, Emma Samuelsson, Christina Åhrén, and Nahid Karami. 2023. "Comparative Genomic Analysis of ST131 Subclade C2 of ESBL-Producing E. coli Isolates from Patients with Recurrent and Sporadic Urinary Tract Infections" Microorganisms 11, no. 7: 1622. https://doi.org/10.3390/microorganisms11071622
APA StyleJaén-Luchoro, D., Kahnamouei, A., Yazdanshenas, S., Lindblom, A., Samuelsson, E., Åhrén, C., & Karami, N. (2023). Comparative Genomic Analysis of ST131 Subclade C2 of ESBL-Producing E. coli Isolates from Patients with Recurrent and Sporadic Urinary Tract Infections. Microorganisms, 11(7), 1622. https://doi.org/10.3390/microorganisms11071622