
Exploring Gut Microbiome Variations between Popillia japonica Populations of Azores
 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection and Processing of Insect and Soil Samples
2.2. Soil and Climatic Parameters
2.3. Next-Generation Sequencing and Processing of Reads
2.4. Metabarcoding and Metagenomic Functional Analysis
3. Results
3.1. Metadata of Amplicon 16S Sequence
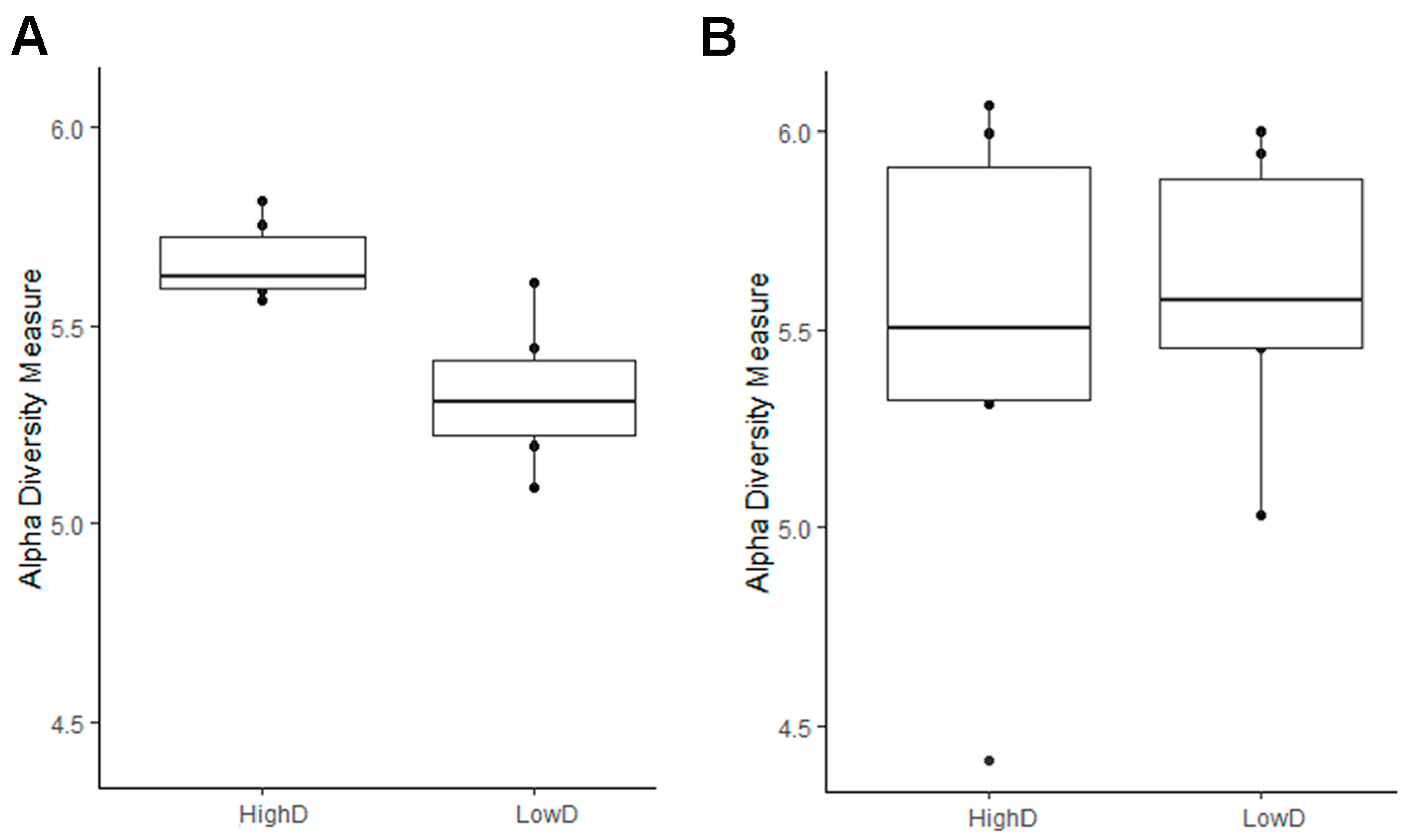
3.2. The Diversity and Composition of the Larval Microbiomes Differ between Insect Populations, While Soil Microbiome Remains Consistent
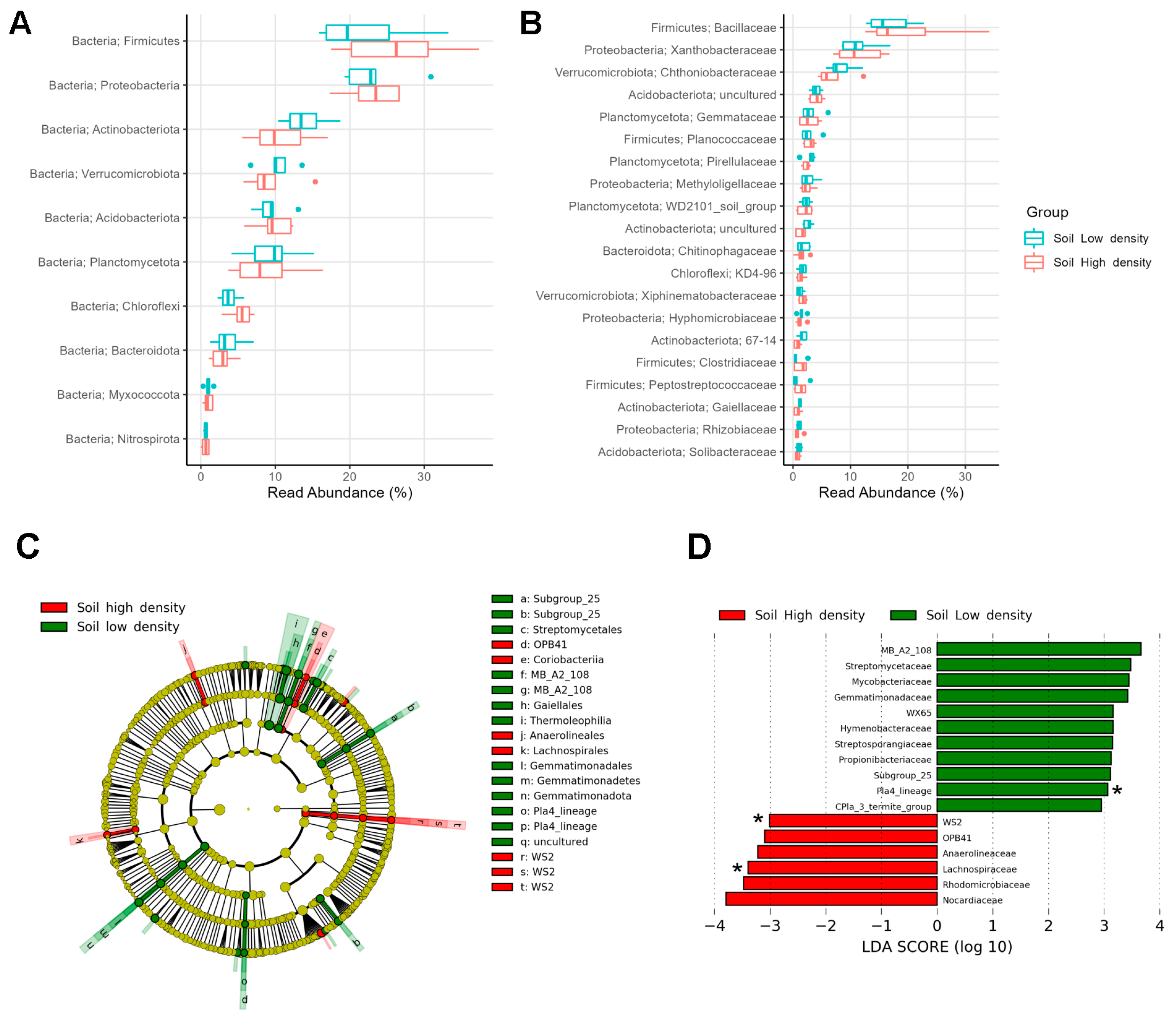
3.3. Soil Microbiomes Revealed Minor Variations between Density Sites
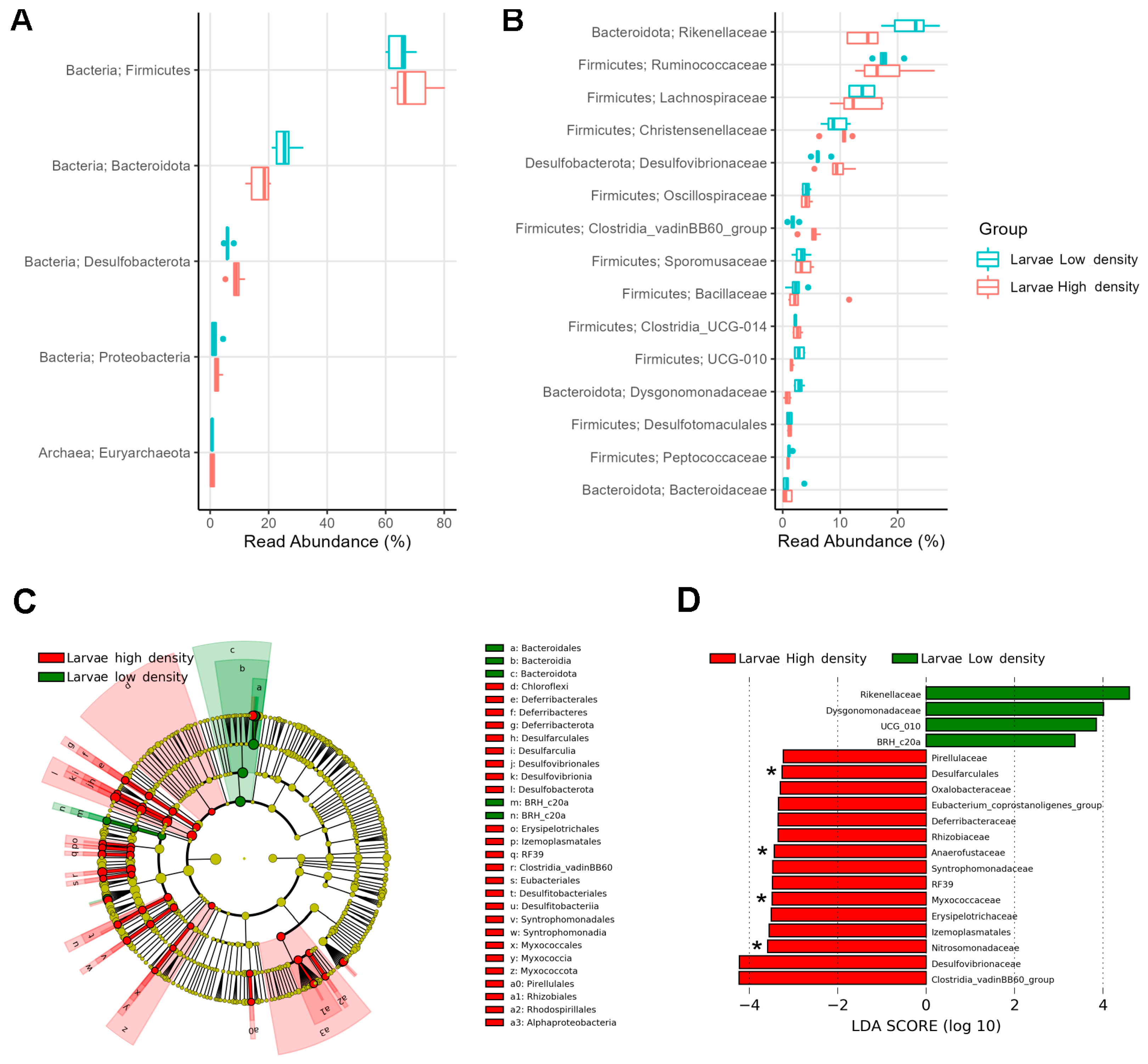
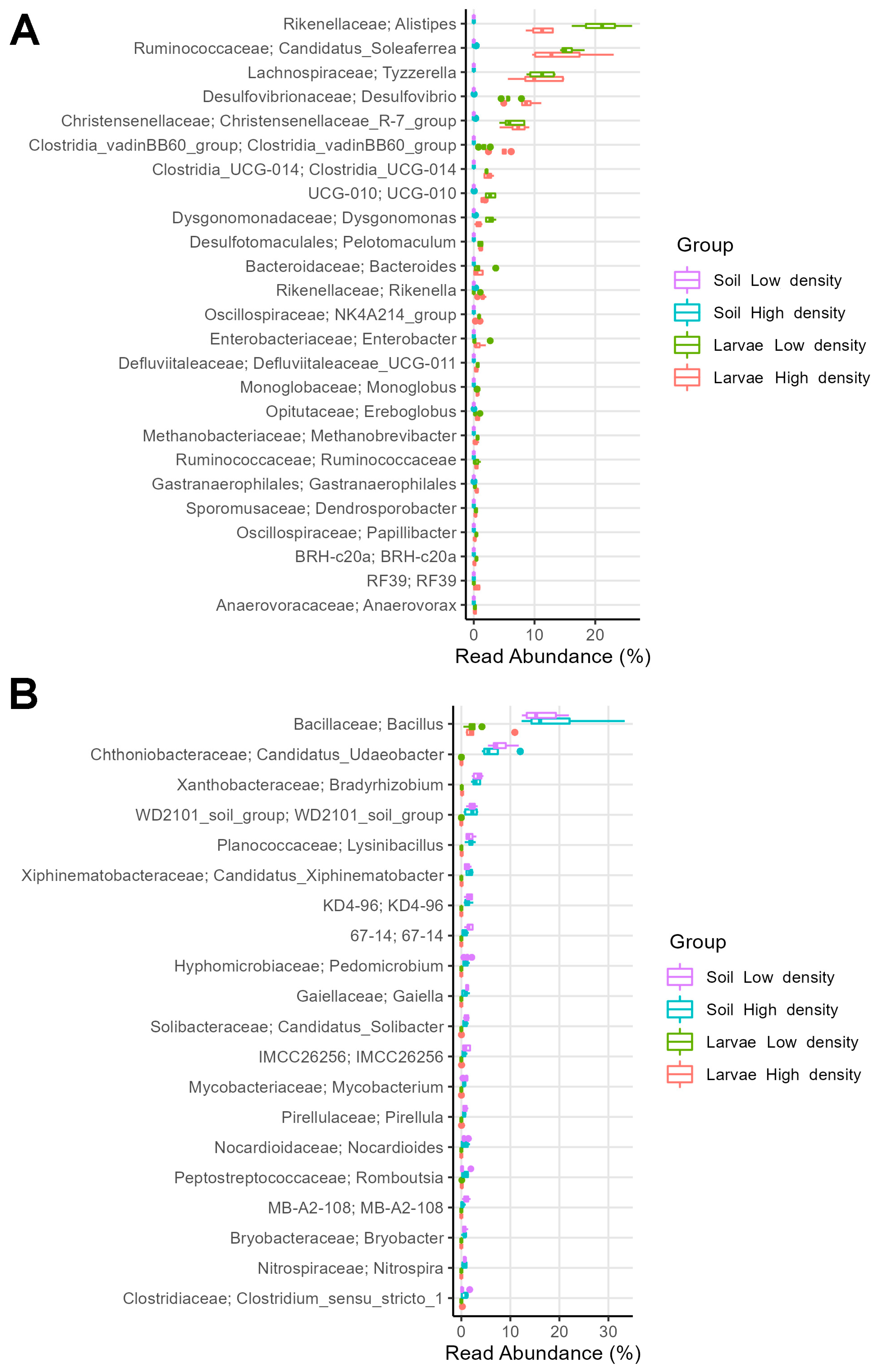
3.4. Some Bacterial Families Were More Enriched in the Microbiome of Larvae from HD than from LD Sites
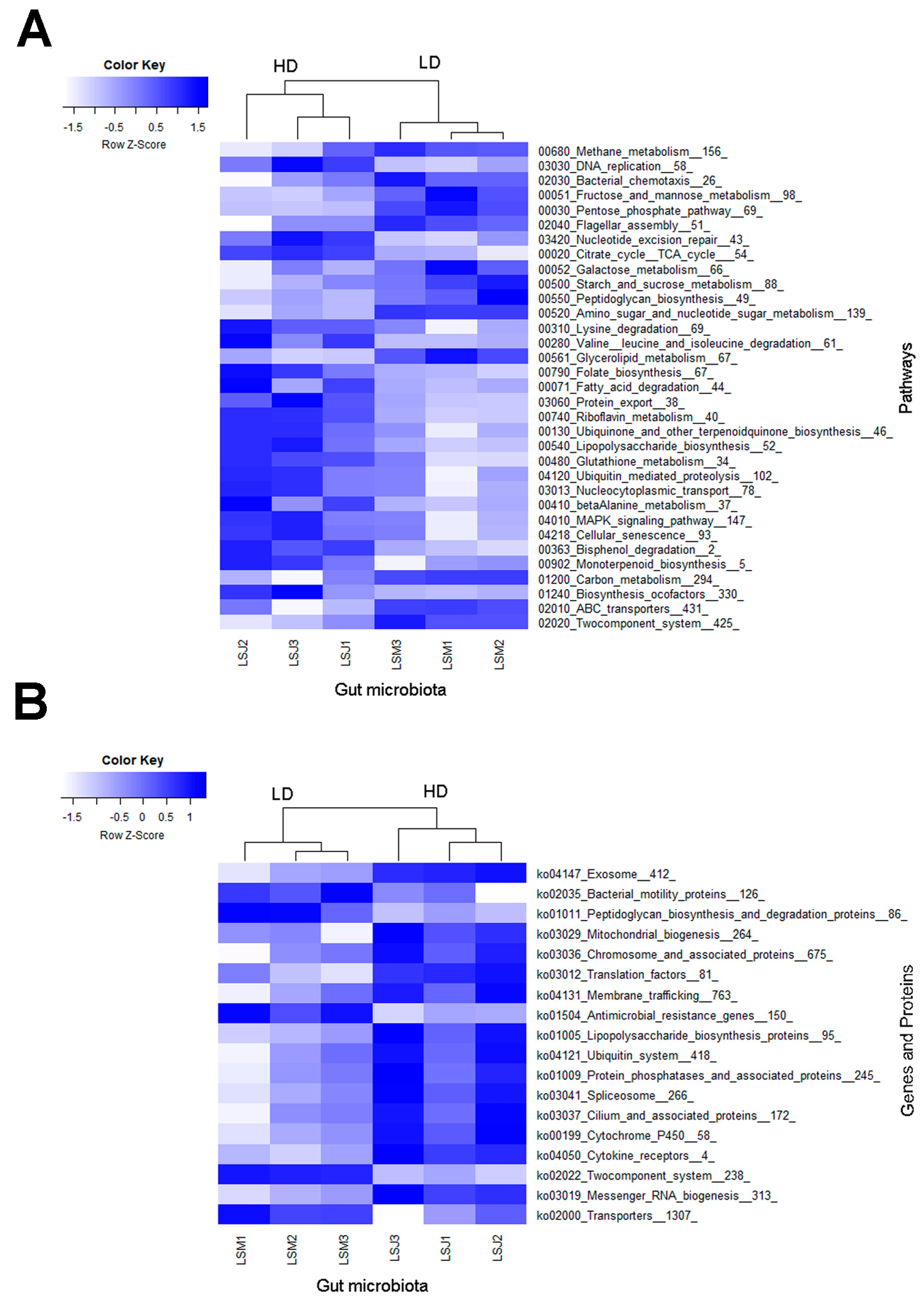
3.5. Metagenomic Analysis Uncovers Enhanced Functional Enrichment in Larvae from the HD Site
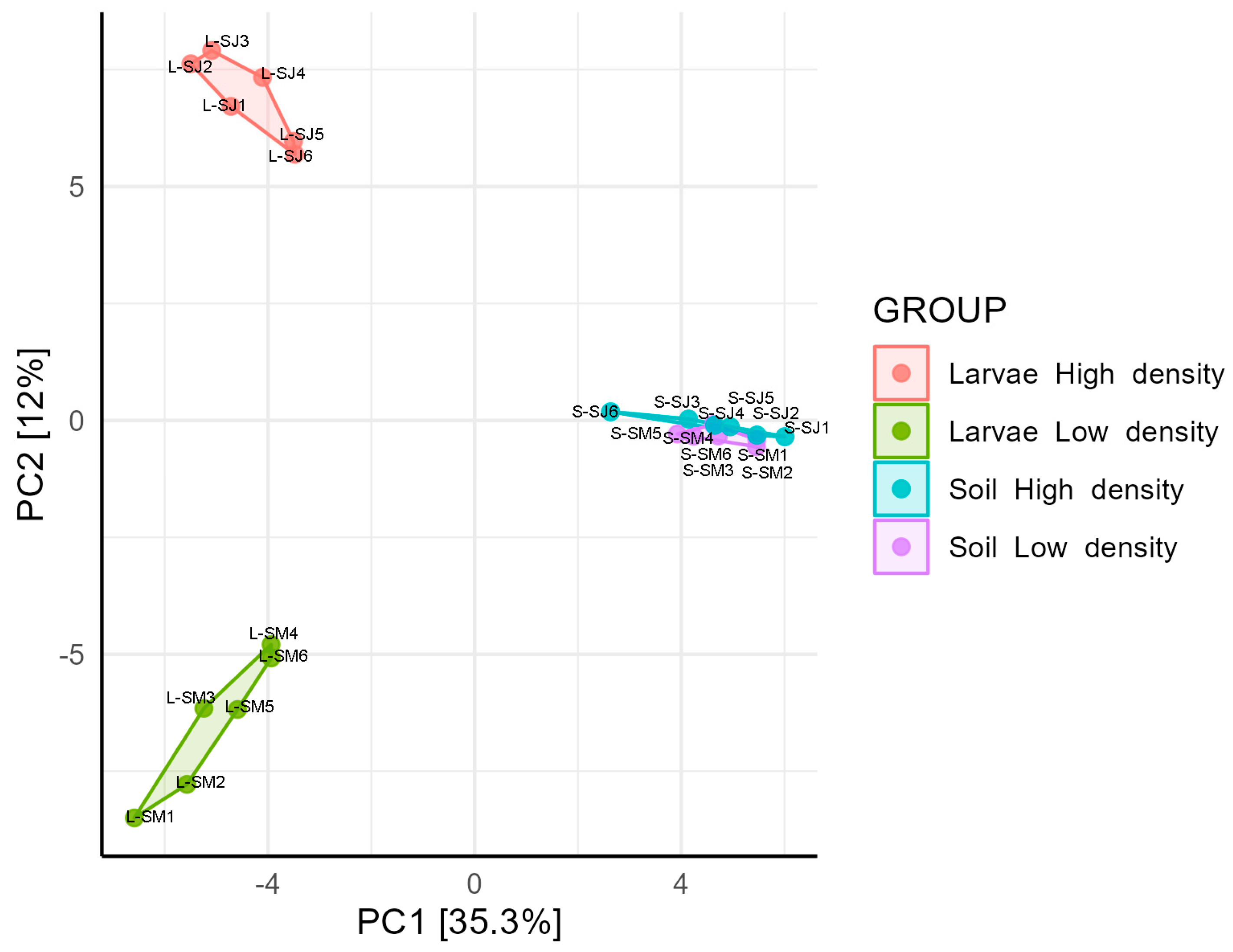
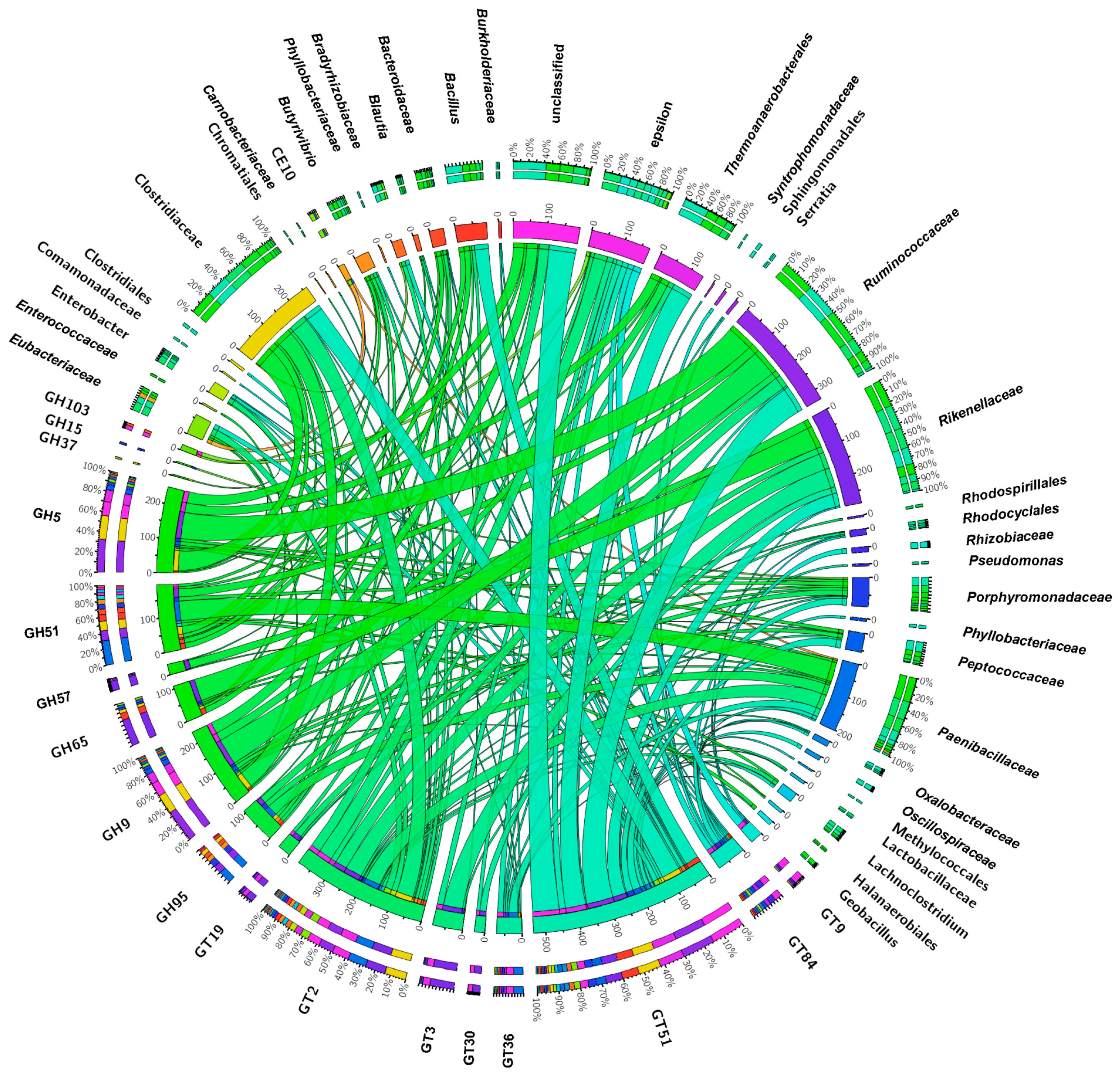
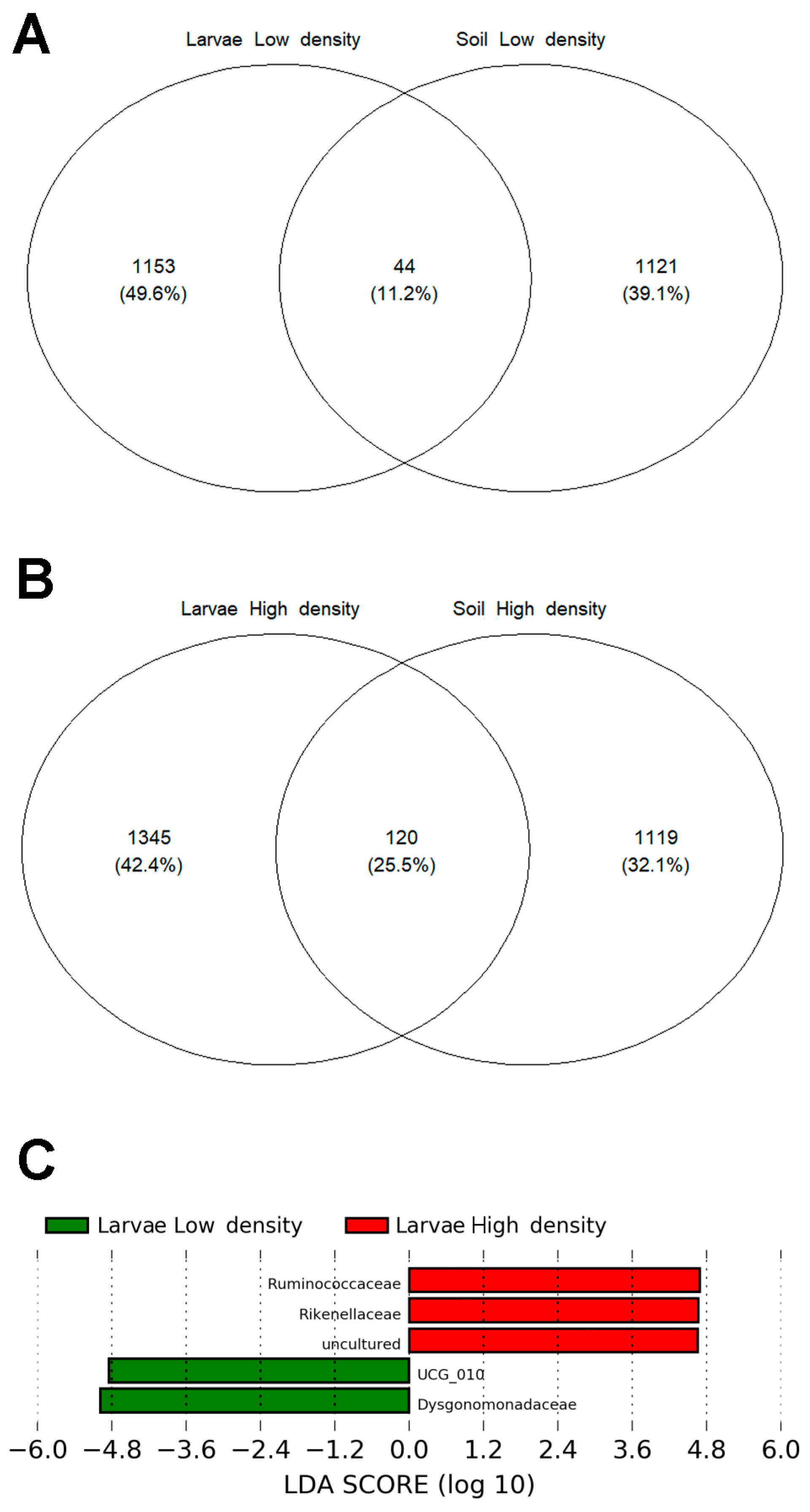
3.6. The Core Microbiome Shared between Soil and Larvae in the HD Site, Is More Diverse and Abundant than in the LD Site
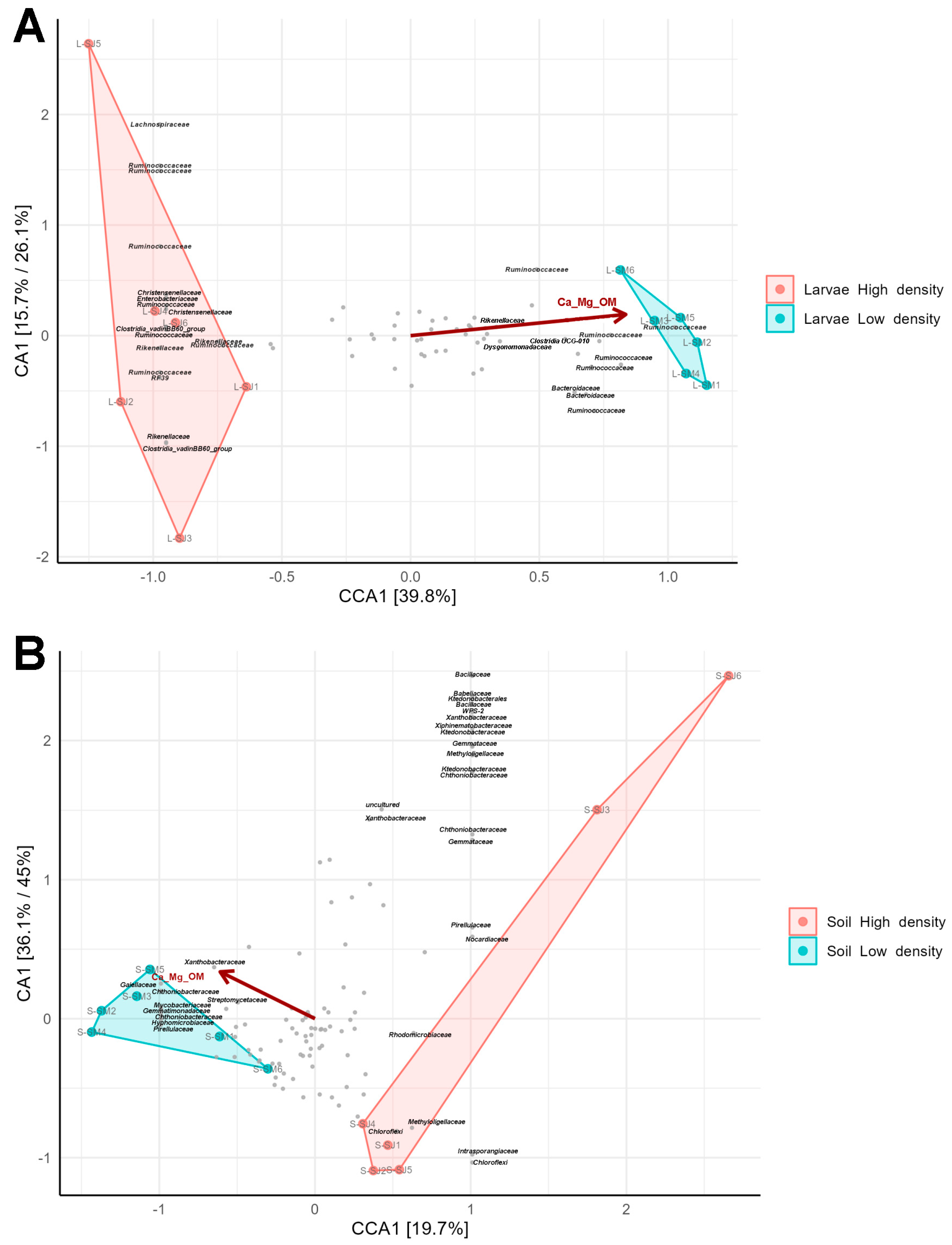
3.7. Microbiome Biomarkers from the Two Density Sites Are Correlated with Soil Parameters
4. Discussion
4.1. The Gut Microbiome of Larvae from HD Sites Is More Favorable to Insect than That of Larvae from LD Sites
4.2. Metagenomic Analysis Uncovers Enhanced Functional Enrichment in the Larvae from the HD Site
4.3. Larvae from the HD Site Share with Soil a Higher Number of Bacteria than Those from LD Site
4.4. There Is a Correlation between Microbiome Biomarkers and Soil Parameters
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Althoff, E.R.; Rice, K.B. Japanese Beetle (Coleoptera: Scarabaeidae) Invasion of North America: History, Ecology, and Management. J. Integr. Pest Manag. 2022, 13, 2. [Google Scholar] [CrossRef]
- Simões, N.; Martins, A. Life Cycle of Popillia Japonica Newman (Coleoptera—Scarabaeidae) in Terceira Island—Azores; University of the Azores: Ponta Delgada, Portugal, 1985. [Google Scholar]
- Martins, A.; Simoes, N. Suppression of the Japanese Beetle in the Azores: An Ecological Approach. Ecol. Bull. 1988, 39, 99–100. [Google Scholar]
- Martins, A.; Paiva, M.R.; Simões, N. Japanese Beetle: Monitoring in the Azores with Semiochemicals. Ecol. Bull. 1988, 39, 101–103. [Google Scholar]
- Simões, N.; Laumond, C.; Bonifassi, E. Effectiveness of Steinernema Spp. and Heterorhabditis Bacteriophora against Popillia Japonica in the Azores. J. Nematol. 1993, 25, 480–485. [Google Scholar]
- Lacey, L.A.; Bettencourt, R.; Garrett, F.J.; Simões, N.J.; Gaugler, R.H. Factors Influencing Parasitism of Adult Japanese Beetles, Polillia Japonica (Col.: Scarabaeidae) by Entomopathogenic Nematodes. Entomophaga 1993, 38, 501–509. [Google Scholar] [CrossRef]
- Simoes, N.; Rosa, J. Survival of Entomophilic Nematodes in Soil [Steinernema Carpocapsae, Steinernema Glaseri]. In Bulletin OILB SROP; OILB: Paris, France, 1994. [Google Scholar]
- Rosa, J.S.; Bonifassi, E.; Amaral, J.; Lacey, L.A.; Simões, N.; Laumond, C. Natural Occurrence of Entomopathogenic Nematodes (Rhabditida: Steinernema, Heterorhabditis) in the Azores. J. Nematol. 2000, 32, 215–222. [Google Scholar] [PubMed]
- Lacey, L.A.; Rosa, J.S.; Simoes, N.O.; Amaral, J.J.; Kaya, H.K. Comparative Dispersal and Larvicidal Activity of Exotic and Azorean Isolates of Entomopathogenic Nematodes against Popillia Japonica (Coleoptera: Scarabaeidae). Eur. J. Entomol. 2001, 98, 439–444. [Google Scholar] [CrossRef] [Green Version]
- Mendes, C.; Lacey, L.; Amaral, J.; Klien, M. Biological Control of Popillia Japonica on Terceira Island (Azores, Portugal): Potential of Bacillus Popilliae. In Bulletin OILB SROP; OILB: Paris, France, 1994. [Google Scholar]
- Popillia Japonica (POPIJA) [Portugal (Azores)]. EPPO Global Database. Available online: https://gd.eppo.int/taxon/POPIJA/distribution/PT_az (accessed on 15 March 2023).
- Smith, L.B. Larval Food Habits of the Japanese Beetle (Popillia Japonica Newm.). J. Econ. Entomol. 1922, 15, 305–310. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N. Embracing the Unknown: Disentangling the Complexities of the Soil Microbiome. Nat. Rev. Microbiol. 2017, 15, 579–590. [Google Scholar] [CrossRef]
- Jansson, J.K.; Hofmockel, K.S. The Soil Microbiome—From Metagenomics to Metaphenomics. Curr. Opin. Microbiol. 2018, 43, 162–168. [Google Scholar] [CrossRef]
- Pineda, A.; Kaplan, I.; Bezemer, T.M. Steering Soil Microbiomes to Suppress Aboveground Insect Pests. Trends Plant Sci. 2017, 22, 770–778. [Google Scholar] [CrossRef] [Green Version]
- Tokuda, G.; Yamaoka, I.; Noda, H. Localization of Symbiotic Clostridia in the Mixed Segment of the Termite Nasutitermes Takasagoensis (Shiraki). Appl. Environ. Microbiol. 2000, 66, 2199–2207. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Segawa, T.; Takeuchi, N.; Barcaza Sepúlveda, G.; Labarca, P.; Kohshima, S.; Hongoh, Y. Metagenomic Analyses Highlight the Symbiotic Association between the Glacier Stonefly Andiperla Willinki and Its Bacterial Gut Community. Environ. Microbiol. 2018, 20, 4170–4183. [Google Scholar] [CrossRef]
- Montagna, M.; Gómez-Zurita, J.; Giorgi, A.; Epis, S.; Lozzia, G.; Bandi, C. Metamicrobiomics in Herbivore Beetles of the Genus Cryptocephalus (Chrysomelidae): Toward the Understanding of Ecological Determinants in Insect Symbiosis. Insect Sci. 2015, 22, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-D.; Li, J.-Y.; Hu, Z.-Q.; Liu, T.-X.; Zhang, S.-Z. Fall Armyworm Gut Bacterial Diversity Associated with Different Developmental Stages, Environmental Habitats, and Diets. Insects 2022, 13, 762. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.G.; Logroño, W.; da Rocha, U.N.; Harms, H.; Nikolausz, M. Enrichment of Anaerobic Microbial Communities from Midgut and Hindgut of Sun Beetle Larvae (Pachnoda Marginata) on Wheat Straw: Effect of Inoculum Preparation. Microorganisms 2022, 10, 761. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Delalibera, I., Jr.; Handelsman, J.; Raffa, K.F. Bacteria Associated with the Guts of Two Wood-Boring Beetles: Anoplophora Glabripennis and Saperda Vestita (Cerambycidae). Environ. Entomol. 2006, 35, 625–629. [Google Scholar] [CrossRef]
- Avila-Arias, H.; Scharf, M.E.; Turco, R.F.; Richmond, D.S. Soil Environments Influence Gut Prokaryotic Communities in the Larvae of the Invasive Japanese Beetle Popillia Japonica Newman. Front. Microbiol. 2022, 13, 854513. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, H. The Impact of Environmental Heterogeneity and Life Stage on the Hindgut Microbiota of Holotrichia Parallela Larvae (Coleoptera: Scarabaeidae). PLoS ONE 2013, 8, e57169. [Google Scholar] [CrossRef]
- Ras, E.; Beukeboom, L.W.; Cáceres, C.; Bourtzis, K. Review of the Role of Gut Microbiota in Mass Rearing of the Olive Fruit Fly, Bactrocera Oleae, and Its Parasitoids. Entomol. Exp. Appl. 2017, 164, 237–256. [Google Scholar] [CrossRef] [Green Version]
- Gurung, K.; Wertheim, B.; Falcao Salles, J. The Microbiome of Pest Insects: It Is Not Just Bacteria. Entomol. Exp. Appl. 2019, 167, 156–170. [Google Scholar] [CrossRef] [Green Version]
- Secretaria Regional da Agricultura e do Desenvolvimento Rural—Secretaria Regional da Agricultura e do Desenvolvimento Rural—Portal. Available online: https://portal.azores.gov.pt/web/sradr (accessed on 28 February 2023).
- Melo, C.D.; Maduro Dias, C.S.A.M.; Wallon, S.; Borba, A.E.S.; Madruga, J.; Borges, P.A.V.; Ferreira, M.T.; Elias, R.B. Influence of Climate Variability and Soil Fertility on the Forage Quality and Productivity in Azorean Pastures. Agriculture 2022, 12, 358. [Google Scholar] [CrossRef]
- Bouyoucos, G.J. The Hydrometer as a New Method for the Mechanical Analysis of Soils. Soil Sci. 1927, 23, 343. [Google Scholar] [CrossRef]
- Andrews, S.; Bittencourt, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics: Cambridge, UK, 2010. [Google Scholar]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Core Team, R. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Andersen, K.S.; Kirkegaard, R.H.; Karst, S.M.; Albertsen, M. Ampvis2: An R Package to Analyse and Visualise 16S RRNA Amplicon Data. BioRxiv 2018. preprint. [Google Scholar]
- Oksanen, F.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.; O’Hara, R.; Simpson, G.; Solymos, P. Vegan: Community Ecology Package; R Package Version 2.4–4; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic Biomarker Discovery and Explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legendre, P.; Gallagher, E.D. Ecologically Meaningful Transformations for Ordination of Species Data. Oecologia 2001, 129, 271–280. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Schmieder, R.; Edwards, R. Fast Identification and Removal of Sequence Contamination from Genomic and Metagenomic Datasets. PLoS ONE 2011, 6, e17288. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. EggNOG-Mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, İ.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An Information Aesthetic for Comparative Genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Chouaia, B.; Goda, N.; Mazza, G.; Alali, S.; Florian, F.; Gionechetti, F.; Callegari, M.; Gonella, E.; Magoga, G.; Fusi, M.; et al. Developmental Stages and Gut Microenvironments Influence Gut Microbiota Dynamics in the Invasive Beetle Popillia Japonica Newman (Coleoptera: Scarabaeidae). Environ. Microbiol. 2019, 21, 4343–4359. [Google Scholar] [CrossRef]
- Kakumanu, M.L.; Maritz, J.M.; Carlton, J.M.; Schal, C. Overlapping Community Compositions of Gut and Fecal Microbiomes in Lab-Reared and Field-Collected German Cockroaches. Appl. Environ. Microbiol. 2018, 84, e01037-18. [Google Scholar] [CrossRef] [Green Version]
- Bauer, E.; Lampert, N.; Mikaelyan, A.; Köhler, T.; Maekawa, K.; Brune, A. Physicochemical Conditions, Metabolites and Community Structure of the Bacterial Microbiota in the Gut of Wood-Feeding Cockroaches (Blaberidae: Panesthiinae). FEMS Microbiol. Ecol. 2015, 91, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Jackson, T.A. Autochthonous Bacterial Flora Indicated by PCR-DGGE of 16S RRNA Gene Fragments from the Alimentary Tract of Costelytra Zealandica (Coleoptera: Scarabaeidae). J. Appl. Microbiol. 2008, 105, 1277–1285. [Google Scholar] [CrossRef]
- Richards, P.; Fothergill, J.; Bernardeau, M.; Wigley, P. Development of the Caecal Microbiota in Three Broiler Breeds. Front. Vet. Sci. 2019, 6, 201. [Google Scholar]
- Hazlewood, G.P.; Gilbert, H.J. Xylan and Cellulose Utilization by the Clostridia. Biotechnol. Read. Mass 1993, 25, 311–341. [Google Scholar]
- Mitchell, W.J. Carbohydrate Assimilation by Saccharolytic Clostridia. Res. Microbiol. 1992, 143, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Rainey, F.A.; Stackebrandt, E. 16S RDNA Analysis Reveals Phylogenetic Diversity among the Polysaccharolytic Clostridia. FEMS Microbiol. Lett. 1993, 113, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Ebert, K.M.; Arnold, W.G.; Ebert, P.R.; Merritt, D.J. Hindgut Microbiota Reflects Different Digestive Strategies in Dung Beetles (Coleoptera: Scarabaeidae: Scarabaeinae). Appl. Environ. Microbiol. 2021, 87, e02100-20. [Google Scholar] [CrossRef]
- Egert, M.; Stingl, U.; Dyhrberg Bruun, L.; Pommerenke, B.; Brune, A.; Friedrich, M.W. Structure and Topology of Microbial Communities in the Major Gut Compartments of Melolontha Melolontha Larvae (Coleoptera: Scarabaeidae). Appl. Environ. Microbiol. 2005, 71, 4556–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dröge, S.; Limper, U.; Emtiazi, F.; Schönig, I.; Pavlus, N.; Drzyzga, O.; Fischer, U.; König, H. In Vitro and in Vivo Sulfate Reduction in the Gut Contents of the Termite Mastotermes Darwiniensis and the Rose-Chafer Pachnoda Marginata. J. Gen. Appl. Microbiol. 2005, 51, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Kuhnigk, T.; Branke, J.; Krekeler, D.; Cypionka, H.; König, H. A Feasible Role of Sulfate-Reducing Bacteria in the Termite Gut. Syst. Appl. Microbiol. 1996, 19, 139–149. [Google Scholar] [CrossRef]
- Schauer, C.; Thompson, C.L.; Brune, A. The Bacterial Community in the Gut of the Cockroach Shelfordella Lateralis Reflects the Close Evolutionary Relatedness of Cockroaches and Termites. Appl. Environ. Microbiol. 2012, 78, 2758–2767. [Google Scholar] [CrossRef] [Green Version]
- Arias-Cordero, E.; Ping, L.; Reichwald, K.; Delb, H.; Platzer, M.; Boland, W. Comparative Evaluation of the Gut Microbiota Associated with the Below- and Above-Ground Life Stages (Larvae and Beetles) of the Forest Cockchafer, Melolontha Hippocastani. PLoS ONE 2012, 7, e51557. [Google Scholar] [CrossRef] [Green Version]
- Gales, A.; Chatellard, L.; Abadie, M.; Bonnafous, A.; Auer, L.; Carrère, H.; Godon, J.-J.; Hernandez-Raquet, G.; Dumas, C. Screening of Phytophagous and Xylophagous Insects Guts Microbiota Abilities to Degrade Lignocellulose in Bioreactor. Front. Microbiol. 2018, 9, 2222. [Google Scholar] [PubMed]
- Chen, J.S. Alcohol Dehydrogenase: Multiplicity and Relatedness in the Solvent-Producing Clostridia. FEMS Microbiol. Rev. 1995, 17, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Johnston, N.C.; Goldfine, H. Phospholipid Aliphatic Chain Composition Modulates Lipid Class Composition, but Not Lipid Asymmetry in Clostridium Butyricum. Biochim. Biophys. Acta 1985, 813, 10–18. [Google Scholar] [CrossRef]
- Jones, D.T.; Woods, D.R. Acetone-Butanol Fermentation Revisited. Microbiol. Rev. 1986, 50, 484–524. [Google Scholar] [CrossRef]
- Elsden, S.R.; Hilton, M.G. Amino Acid Utilization Patterns in Clostridial Taxonomy. Arch. Microbiol. 1979, 123, 137–141. [Google Scholar] [CrossRef]
- Prosser, J.I.; Head, I.M.; Stein, L.Y. The Family Nitrosomonadaceae. In The Prokaryotes: Alphaproteobacteria and Betaproteobacteria; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 901–918. ISBN 978-3-642-30197-1. [Google Scholar]
- Caballero, S.; Galeano, A.M.; Lozano, J.D.; Vives, M. Description of the Microbiota in Epidermal Mucus and Skin of Sharks (Ginglymostoma Cirratum and Negaprion Brevirostris) and One Stingray (Hypanus Americanus). PeerJ 2020, 8, e10240. [Google Scholar] [CrossRef]
- Neis, E.P.J.G.; Dejong, C.H.C.; Rensen, S.S. The Role of Microbial Amino Acid Metabolism in Host Metabolism. Nutrients 2015, 7, 2930–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uebanso, T.; Shimohata, T.; Mawatari, K.; Takahashi, A. Functional Roles of B-Vitamins in the Gut and Gut Microbiome. Mol. Nutr. Food Res. 2020, 64, e2000426. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, K.; Hosomi, K.; Sawane, K.; Kunisawa, J. Metabolism of Dietary and Microbial Vitamin B Family in the Regulation of Host Immunity. Front. Nutr. 2019, 6, 48. [Google Scholar]
- Zug, R.; Hammerstein, P. Wolbachia and the Insect Immune System: What Reactive Oxygen Species Can Tell Us about the Mechanisms of Wolbachia–Host Interactions. Front. Microbiol. 2015, 6, 1201. [Google Scholar] [PubMed] [Green Version]
- Hirakawa, H.; Kurushima, J.; Hashimoto, Y.; Tomita, H. Progress Overview of Bacterial Two-Component Regulatory Systems as Potential Targets for Antimicrobial Chemotherapy. Antibiotics 2020, 9, 635. [Google Scholar] [CrossRef] [PubMed]
- Carmody, R.N.; Turnbaugh, P.J. Host-Microbial Interactions in the Metabolism of Therapeutic and Diet-Derived Xenobiotics. J. Clin. Investig. 2014, 124, 4173–4181. [Google Scholar] [CrossRef] [Green Version]
- Kelly, S.L.; Kelly, D.E. Microbial Cytochromes P450: Biodiversity and Biotechnology. Where Do Cytochromes P450 Come from, What Do They Do and What Can They Do for Us? Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120476. [Google Scholar] [CrossRef] [Green Version]
- Jackson, D.N.; Theiss, A.L. Gut Bacteria Signaling to Mitochondria in Intestinal Inflammation and Cancer. Gut Microbes 2020, 11, 285–304. [Google Scholar] [CrossRef] [Green Version]
- Högbom, M.; Ihalin, R. Functional and Structural Characteristics of Bacterial Proteins That Bind Host Cytokines. Virulence 2017, 8, 1592–1601. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, Ó.; Fernández-Fernández, M.; Valls, C.; Valenzuela, S.V.; Roncero, M.B.; Vidal, T.; Díaz, P.; Pastor, F.I.J. Characterization of a Family GH5 Xylanase with Activity on Neutral Oligosaccharides and Evaluation as a Pulp Bleaching Aid. Appl. Environ. Microbiol. 2010, 76, 6290. [Google Scholar] [CrossRef] [Green Version]
- Numan, M.T.; Bhosle, N.B. α-l-Arabinofuranosidases: The Potential Applications in Biotechnology. J. Ind. Microbiol. Biotechnol. 2006, 33, 247–260. [Google Scholar] [CrossRef]
- Aymé, L.; Hébert, A.; Henrissat, B.; Lombard, V.; Franche, N.; Perret, S.; Jourdier, E.; Heiss-Blanquet, S. Characterization of Three Bacterial Glycoside Hydrolase Family 9 Endoglucanases with Different Modular Architectures Isolated from a Compost Metagenome. Biochim. Biophys. Acta BBA—Gen. Subj. 2021, 1865, 129848. [Google Scholar] [CrossRef]
- Nihira, T.; Nishimoto, M.; Nakai, H.; Ohtsubo, K.; Kitaoka, M. Characterization of Two α-1,3-Glucoside Phosphorylases from Clostridium Phytofermentans. J. Appl. Glycosci. 2014, 61, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Nagare, M.; Ayachit, M.; Agnihotri, A.; Schwab, W.; Joshi, R. Glycosyltransferases: The Multifaceted Enzymatic Regulator in Insects. Insect Mol. Biol. 2021, 30, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Andert, J.; Marten, A.; Brandl, R.; Brune, A. Inter- and Intraspecific Comparison of the Bacterial Assemblages in the Hindgut of Humivorous Scarab Beetle Larvae (Pachnoda Spp.). FEMS Microbiol. Ecol. 2010, 74, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Ceja-Navarro, J.A.; Vega, F.E.; Karaoz, U.; Hao, Z.; Jenkins, S.; Lim, H.C.; Kosina, P.; Infante, F.; Northen, T.R.; Brodie, E.L. Gut Microbiota Mediate Caffeine Detoxification in the Primary Insect Pest of Coffee. Nat. Commun. 2015, 6, 7618. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frias, J.; Garriga, A.; Peñalver, Á.; Teixeira, M.; Beltrí, R.; Toubarro, D.; Simões, N. Exploring Gut Microbiome Variations between Popillia japonica Populations of Azores. Microorganisms 2023, 11, 1972. https://doi.org/10.3390/microorganisms11081972
Frias J, Garriga A, Peñalver Á, Teixeira M, Beltrí R, Toubarro D, Simões N. Exploring Gut Microbiome Variations between Popillia japonica Populations of Azores. Microorganisms. 2023; 11(8):1972. https://doi.org/10.3390/microorganisms11081972
Chicago/Turabian StyleFrias, Jorge, Anna Garriga, Ángel Peñalver, Mário Teixeira, Rubén Beltrí, Duarte Toubarro, and Nelson Simões. 2023. "Exploring Gut Microbiome Variations between Popillia japonica Populations of Azores" Microorganisms 11, no. 8: 1972. https://doi.org/10.3390/microorganisms11081972